





Abstract
Background的evolution in pulmonary arterial hypertension (PAH) management has been summarised in three iterations of the European Society of Cardiology/European Respiratory Society (ESC/ERS) guidelines. No study has assessed whether changes in management, as reflected in the changing guidelines, has translated to improved long-term survival in PAH.
MethodsWe performed a mixed retrospective/prospective analysis of treatment-naïve, incident PAH patients (n=392) diagnosed at three major centres in Canada from 2009 to 2021. Patients were divided into two groups based on their diagnosis date and in accordance with the 2009 and 2015 ESC/ERS guideline iterations. Overall survival was assessed based on date of diagnosis and initial treatment strategy (i.e.monotherapyversuscombination therapy).
Results的re was a shift towards more aggressive upfront management with combination therapy in Canada after the publication of the 2015 ESC/ERS guidelines (10.4% and 30.8% in patients from 2009 to 2015 and 36.0% and 57.4% in patients diagnosed after 2015 for baseline and 2-year follow-up, respectively). A key factor associated with combination therapy after 2015 was higher pulmonary vascular resistance (p=0.009). The 1-, 3- and 5-year survival rates in Canada were 89.2%, 75.6% and 56.0%, respectively. Despite changes in management, there was no improvement in long-term survival before and after publication of the 2015 ESC/ERS guidelines (p=0.53).
Conclusions的re was an increase in the use of initial and sequential combination therapy in Canada after publication of the 2015 ESC/ERS guidelines, which was not associated with improved long-term survival. These data highlight the continued difficulties of managing this aggressive pulmonary disease in an era without a cure.
Abstract
Incident PAH patients in Canada have been treated more aggressively in recent years, reflecting changing ESC/ERS guideline recommendations, but this was not mirrored by incremental improvement in survival since 2015https://bit.ly/3lyc1Oc
Introduction
Pulmonary arterial hypertension (PAH) is a progressive pulmonary arteriopathy, which engenders elevations in pulmonary vascular resistance (PVR) and right ventricular strain [1,2]. Right heart failure is a common sequel of PAH and represents the leading cause of death in these patients [3]. Over the last 40 years, a number of national registries have yielded valuable epidemiological data on demographics, haemodynamics, prognostic factors and real-world treatment approaches for patients with PAH. As these registries cumulatively span several decades, they offer unique insights into secular trends in PAH mortality.
的natural progression of the disease in the pre-treatment era of PAH was captured by the US National Institutes of Health (NIH) Primary Pulmonary Hypertension Registry in the 1980s [3]. At this time, outcomes were grim, with 1- and 5-year survival rates of only 68% and 34%, respectively [3]. The advent of epoprostenol in the early 1990s revolutionised treatment for these patients, conferring marked improvements in survival, with 1- and 5-year survival rates of 88% and 47%, respectively (∼14–20% absolute improvement) [4]. Since the introduction of epoprostenol, five main classes of pharmaceutical agents have been approved for the treatment of PAH. PAH treatment options now include prostacyclin analogues/prostacyclin receptor agonists, endothelin receptor antagonists and phosphodiesterase-5 inhibitors/activators of soluble guanylate cyclase, which target three aberrant pathways: prostacyclin, endothelial and nitric oxide pathways, respectively. The primary mechanism of action for these therapies relates to vasodilation with subsequent improvement in symptoms and improved functional capacity, but their ability to prevent disease progression and improve survival remains less certain [5].
的evolution in PAH management is reflected in several iterations of the American [6–8], Canadian [9] and European guidelines [10–12]. One of the most significant advancements in PAH management was reflected in the 2015 joint European Society of Cardiology/European Respiratory Society (ESC/ERS) clinical guidelines, which introduced the concept of initial risk-guided therapy [10]. Treatment algorithms were incorporated into these contemporary guidelines, illustrating an important shift towards more aggressive upfront management with combination therapy. These changes were subsequently endorsed by the 6th World Symposium on Pulmonary Hypertension in 2018 [13]. That is, for patients deemed “high risk”, initial combination therapy including intravenous prostacyclin is recommended. Conversely, guidelines recommend patients deemed “low risk” or “intermediate risk” be treated with either initial monotherapy or dual oral combination therapy. To date, no study has evaluated whether these episodic changes in guideline treatment recommendations were followed by incremental improvements in long-term survival in a real-world population.
的re is a paucity of multicentre Canadian PAH outcomes data as the first prospective clinical Canadian registry was only recently initiated. As such, we have little insight into the changing landscape of PAH disease characteristics and mortality across Canada. Therefore, the aims of the present study were twofold: 1) to describe any temporal changes in demographics, disease characteristics and management of PAH in Canada, and 2) to describe the secular trends in PAH mortality in Canada. We hypothesised that there would be increasing use of combination therapy and improvements in mortality following major PAH treatment guideline updates, particularly after 2015.
Methods
This study was approved by the Research Ethics Review Board at the University of Ottawa Heart Institute (Ottawa, ON) (20180423-01H), University of Calgary (Calgary, AB) (REB20-0916) and University of British Columbia (Vancouver, BC) (H20-01322).
Canadian PAH cohort
This analysis includes a cohort of treatment-naïve World Health Organization (WHO) Group 1 PAH patients diagnosed at three major PAH centres in Canada: Ottawa, Calgary and Vancouver. Group 1 PAH was ascertained by right heart catheterisation (RHC) in accordance with clinical guidelines at the time of diagnosis [14]. At diagnosis, patient demographics, disease characteristics and haemodynamic data were recorded.
Patient PAH-specific therapies were also documented, specifically whether they were placed on initial monotherapy (i.e.one PAH agent) or combination therapy (i.e.dual or triple therapy). A patient's initial therapy was defined as treatment up to 3 months post-diagnosis. To determine temporal changes in management, the patient's PAH therapy at 2 years post-diagnosis, death or last clinical visit was recorded.
Ottawa
Treatment-naïve WHO Group 1 PAH patients who received their incident diagnosis at the University of Ottawa Heart Institute between 1 January 2009 and 30 October 2017 were retrospectively identified. After 30 October 2017, patients were prospectively enrolled until 20 August 2021.
Calgary
Patients diagnosed from January 2015 until April 2018 were retrospectively identified from a local RHC database. After April 2018, patients were enrolled prospectively in the Canadian Pulmonary Hypertension Registry using PAHTool (INOVULTUS, Santa Maria da Feira, Portugal) until March 2021.
Vancouver
Patients were prospectively enrolled in the Canadian Pulmonary Hypertension Registry from 1 January 2017 until 24 January 2021.
Treatment era
Patients were divided into two groups according to their incident diagnosis date and in relation to iterations of the ESC/ERS PAH treatment guidelines: 1) 2009–2015 (2009 update) and 2) August 2015 to present (2015 update) (table 1).
European Society of Cardiology/European Respiratory Society (ESC/ERS) guidelines for initial therapy for nonresponders to acute vasoreactivity testing
Statistical analysis
Differences in demographics and disease characteristics between treatment eras were compared using ANOVA or the Chi-squared test where appropriate. The primary end-point was transplant-free survival. All patients were followed until death, lung or heart–lung transplant, or their last clinical encounter, whichever occurred first. Patients were censored at the end of the follow-up period. Transplant-free survival was analysed using the Kaplan–Meier method and differences between risk strata were assessed by the log-rank test.
To examine the impact of initial therapy on survival in patients diagnosed after 2015, a propensity score was first developed to account for the selection bias and nonrandomised treatment allocation of monotherapyversuscombination therapy. Specifically, this approach used a logistic regression model to summarise measured covariates that were predictors of this decision into a single composite score that represents a probability of a patient being treated with monotherapyversuscombination therapy. For this analysis, missing values of the covariates were imputed (multiple imputations) to ensure that propensity scores could be calculated for all patients. The distribution of the propensity scores derived from the multiple imputations were compared between patients who were placed on monotherapy and combination therapy. A total of 14 demographic and clinical covariates that were associated with initial treatment strategy were entered into the propensity score model. Next, we used a multivariable Cox proportional hazards model to assess for the hazard ratio (HR) associated with combination therapy compared with patients initiated on monotherapy therapy; this was adjusted for the propensity score, New York Heart Association (NYHA) Function Class, PVR and ESC/ERS risk status. A p-value <0.05 was considered statistically significant. Statistical analyses were performed using SAS version 9.4 (SAS Institute, Cary, NC, USA) and graphics were created using the R statistical environment (www.r-project.org).Code is available upon request.
Results
Demographic characteristics at diagnosis
我们发现了一个群435事件和treatment-naïve WHO Group 1 PAH patients in three major cities across Canada: Ottawa (n=234), Calgary (n=111) and Vancouver (n=47). As expected, this cohort was comprised of predominantly idiopathic PAH (IPAH) and PAH associated with connective tissue disease, at 58.4% and 30.4%, respectively (table 2).At the time of diagnosis, patients had a mean±sd61.1±16.4岁,绝大多数(64.0%)experiencing NYHA Functional Class III symptoms. Incident RHC revealed a mean pulmonary arterial pressure (mPAP) of 45.2±12.7 mmHg, mean right atrial pressure (mRAP) of 8.5±5.4 mmHg, PVR of 9.7±5.3 WU and cardiac index of 2.2±0.7 L·min−1·m−2.
Patient demographic and disease characteristics separated by diagnosis date and in accordance with publication of the European Society of Cardiology/European Respiratory Society (ESC/ERS) guidelines
Temporal changes in patient demographics
Patients were grouped relative to their diagnosis date and in accordance with publication of the respective ESC/ERS guideline iteration (i.e.2009–2015 or after 2015). Baseline demographics and comorbidities were similar over the 12-year follow-up period (table 2).After 2015, there was a slight decrease in the proportion of patients diagnosed with IPAH (64% in 2009–2015versus56%, 2015),博士的比例增加ug/toxin-induced PAH and portal hypertension (table 2).RHC data also revealed a similar degree of PAH severity over this time period, with no differences in mPAP, mRAP, PVR or cardiac index at diagnosis (table 2).
Initial PAH therapy
治疗的进步在过去的二十年中have been summarised in multiple iterations of the ESC/ERS guidelines (table 1).在2015年的更新,支持switc指导方针h from NYHA Functional Class to a risk-based therapeutic strategy. In our Canadian cohort of incident PAH patients, the NYHA Functional Class and ESC/ERS risk status were similar over the course of the two guideline implementation periods (figure 1band d). As expected, patients presenting with advanced NYHA Functional Class (figure 1a) or ESC/ERS risk status (figure 1c) had a higher risk of mortality (p<0.001).
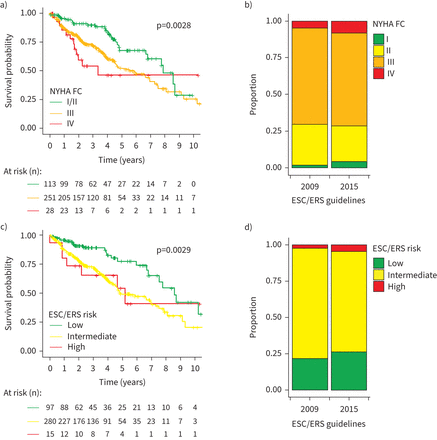
Kaplan–Meier curves for baseline a) New York Heart Association (NYHA) Functional Class (FC) and c) European Society of Cardiology/European Respiratory Society (ESC/ERS)-based risk assessment, and b, d) the associating proportion of patients per ESC/ERS guideline iteration.
Over time, there has been a transition to more aggressive upfront management with dual or even triple therapy for patients at elevated risk. Between 2009 and 2015, 85% of patients were treated with initial monotherapy (figure 2a), with the majority being prescribed bosentan or tadalafil (figure 2b).After 2015, 40.1% of patients who were not vasoreactive and qualified for treatment (NYHA Functional Class II–IV) with PAH therapies were placed on initial combination therapy (figure 2c).In patients with NYHA Functional Class III/IV symptoms, 45.0% were placed on either initial dual or triple therapy (figure 2c).的percentage of patients on combination therapy at 2-year follow-up further improved, from only 30.8% of patients diagnosed from 2009 to 2015 to 57.4% in patients diagnosed after 2015 (figure 2a).的demographics and disease characteristics of patients initiated on combinationversusmonotherapy, and who were diagnosed after 2015, are illustrated insupplementary table S1. Patients placed on initial combination therapy were, on average, younger (57.8±16.6versus63.1±15.9 years; p=0.013) and tended to have a lower burden of comorbidities such as diabetes (monotherapy 35.2%versuscombination therapy 20.8%; p=0.029) and atrial fibrillation (monotherapy 21.4%versuscombination therapy 11.5%; p=0.08). Patients placed on combination therapy also had a higher proportion of NYHA Functional Class III/IV symptoms (p=0.024) and had more severe haemodynamics (PVR 11.9±5.8versus8.4±4.4 WU; p<0.0001 and cardiac index 2.0±0.6versus2.3±0.6 L·min−1·m−2; p=0.0027).
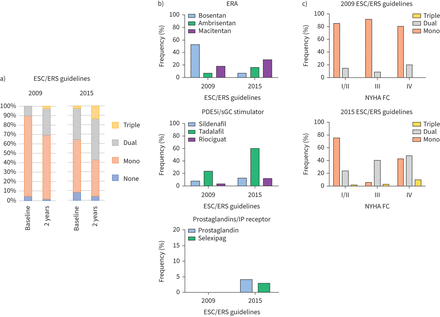
a) Class of initial and follow-up pulmonary arterial hypertension (PAH) therapy per European Society of Cardiology/European Respiratory Society (ESC/ERS) guideline iteration. This was further divided according to b) PAH-specific agents and c) New York Heart Association (NYHA) Functional Class (FC). ERA: endothelin receptor antagonist; PDE5i: phosphodiesterase type 5 inhibitor; sGC: soluble guanylate cyclase; IP: prostacyclin.
Survival
的median (interquartile range) follow-up was 2.91 (1.45–4.58) years with a maximum follow-up duration of 11.7 years. During this time period there were 143 deaths and four lung transplants. The 1-, 3- and 5-year survival rates in our Canadian PAH cohort were 89.2% (95% CI 86.2–92.4%), 75.6% (95% CI 71.1–80.4%) and 56.0% (95% CI 49.9–62.8%), respectively. In the overall population, there was no impact of diagnosis date on 1- or 5-year mortality rates (figure 3).Specifically, 1-year survival rates for patients diagnosed during the 2009 and 2015 guideline eras were 88.8% and 89.5%, respectively (p=0.87). Similarly, the 5-year survival rates were also not statistically different across these time periods (53.8% and 58.9%, respectively; p=0.53). In our cohort, age at diagnosis (p<0.0001) (supplementary figure S1) and male sex (p=0.0035) (supplementary figure S2)与更高的死亡风险。的lack of temporal improvement in survival across the two guideline iterations was consistent even when stratifying the cohort by age (supplementary figure S1), sex (supplementary figure S2) and PAH subtype (supplementary figure S3).
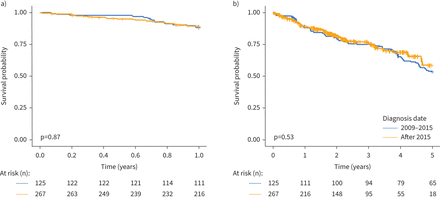
European Society of Cardiology/European Respiratory Society (ESC/ERS) treatment era (2009–2015 and after 2015) and survival in pulmonary arterial hypertension patients: a) 1-year and b) 5-year post-diagnosis survival was not different before and after publication of the 2015 ESC/ERS guidelines.
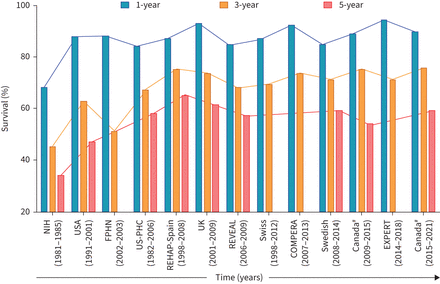
Survival rates from historical and contemporary PAH registries were graphed in chronological order (figure 4).In our Canadian PAH cohort, survival rates were comparable to other time-matched national registries. After the introduction of epoprostenol in the early 1990s, 1-year survival rates appear to have plateaued for the subsequent three decades. The 3- and 5-year survival rates reached their zenith in the early 2000s, and seem to have similarly plateaued for the successive two decades.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
![Survival of incident patients in historical and contemporary pulmonary arterial hypertension (PAH) registries: 1-, 3- and 5- year survival rates are displayed with registries placed in chronological order. NIH: National Institutes of Health Pulmonary Hypertension Registry [3]; USA: McLaughlin et al. [4]; FPHN: French Pulmonary Hypertension Registry [26]; US-PHC: Pulmonary Hypertension Connection database [27]; REHAP-Spain: Spanish Registry of PAH [28]; UK: UK PAH Registry [29]; REVEAL: Registry to Evaluate Early And Long-term PAH Disease Management [30]; Swiss: Swiss PAH Registry [31]; COMPERA: European PAH Registry (Comparative, Prospective Registry of Newly Initiated Therapies for Pulmonary Hypertension) [32]; Swedish: Swedish PAH Registry [33]; EXPERT: Exposure Registry Riociguat in Patients with Pulmonary Hypertension [34]. #: data from our Canadian PAH cohort of treatment-naïve Group 1 PAH patients.](http://www.qdcxjkg.com/content/erj/59/6/2101552/F4.large.jpg?width=800&height=600&carousel=1){kind=link}
{kind=link}
Survival of incident patients in historical and contemporary pulmonary arterial hypertension (PAH) registries: 1-, 3- and 5- year survival rates are displayed with registries placed in chronological order. NIH: National Institutes of Health Pulmonary Hypertension Registry [3];USA: McLaughlinet al.[4];FPHN: French Pulmonary Hypertension Registry [26];US-PHC: Pulmonary Hypertension Connection database [27];REHAP-Spain: Spanish Registry of PAH [28];UK: UK PAH Registry [29];REVEAL: Registry to Evaluate Early And Long-term PAH Disease Management [30];Swiss: Swiss PAH Registry [31];COMPERA: European PAH Registry (Comparative, Prospective Registry of Newly Initiated Therapies for Pulmonary Hypertension) [32];Swedish: Swedish PAH Registry [33];专家:暴露注册Riociguat病人with Pulmonary Hypertension [34].#: data from our Canadian PAH cohort of treatment-naïve Group 1 PAH patients.
Initial treatment strategy and survival
In the overall population, there was no difference in 1- or 5-year (figure 5aand b) survival in patients who received upfront monotherapy (5-year, 52.7%) or dual therapy (5-year, 59.2%; p=0.32). There was also no difference in survival between patients on sequential combination therapy and patients placed on initial dual therapy (p=0.45) (supplementary figure S4).In an exploratory analysis, there was an early signal that upfront triple therapy may confer a survival benefit (figure 5); however, there was likely insufficient power to detect a difference (n=7).
{kind=link}
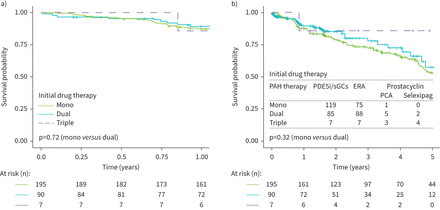
{kind=link}
Survival and initial drug therapy: a) 1-year and b) 5-year post-diagnosis survival according to initial treatment with a single agent (mono), dual or triple therapy. PDE5i: phosphodiesterase type 5 inhibitor; sGCs: soluble guanylate cyclase stimulator; ERA: endothelin receptor antagonist; PCA: prostacyclin analogue.
的largest trial in support of upfront combination therapy was the AMBITION (Ambrisentan and Tadalafil in Patients with Pulmonary Arterial Hypertension) trial, which compared ambrisentan and tadalafilversuseither drug alone as monotherapy [15]. When the inclusion/exclusion criteria were applied to our Canadian cohort, only 41.8% would have qualified (supplementary figure S5).In our Canadian cohort, this trial would have preselected a group of patients with a more favourable prognosis (p=0.0002).
Multivariable modelling
Overall population
的re was no association between upfront combination therapy and better survival after adjusting for baseline ESC/ERS risk category (dual therapyversusmonotherapy HR 0.70, 95% CI 0.45–1.08; p=0.11).
Diagnosis date after 2015: logistic regression-derived propensity score
Logistic regression identified multiple factors as predictive of being treated with initial combination therapy in patients receiving a PAH diagnosis after 2015 (χ2=43.2, C-index=0.75) (table 3).In the multivariable analysis, PVR (χ2=6.76; p=0.0098) and age at diagnosis (χ2=2.10; p=0.15) were the predominant factors associated with use of initial combination therapy (table 3).的predicted likelihood of combination therapy for each individual patient was determined from this model and was entered into the Cox proportional hazards model as a propensity score to adjust for the lack of randomisation (table 4).In the Cox model, there was also no association between initial combination therapy and survival after adjusting for the propensity score, PVR and ESC/ERS risk status (HR 0.76, 95% CI 0.40–1.46).
倾向分数:物流regressi的结果on modelling for initial combination therapy (overall model χ2=43.2, C-index=0.75)
Cox proportional hazards model for all-cause death
Discussion
Data derived from major historical and contemporary PAH registries demonstrate only modest improvements in short- and long-term survival over the past few decades despite several guideline iterations and new drugs that target the three key pathways. This study investigated secular trends in mortality in patients with PAH in Canada, focusing on changes after the publication of the 2015 ESC/ERS guidelines. Major findings from our Canadian cohort were: 1) the demographics and pulmonary haemodynamics of PAH patients were stable over the 12-year follow-up period, 2) a higher proportion of patients were treated with initial dual or triple therapy after publication of the 2015 ESC/ERS guidelines, 3) despite clear evolution in PAH management strategies, there was no parallel improvement in 1- or 5-year survival rates at three major PAH centres in Canada after publication of the 2015 guidelines, and 4) initial dual therapy was not associated with better transplant-free survival compared with initial monotherapy. Together, this body of work highlights the difficulties of managing this aggressive pulmonary vascular disease in an era without a cure.
To the best of our knowledge, this is the first study to examine long-term survival before and after publication of the 2015 ESC/ERS guidelines. A key finding from our study was an absence of improvement in survival despite clear efforts to comply with the prevailing guidelines. These data add credence to our understanding that contemporary management does not target causative mechanisms and may fail to improve overall survival. Our data is supported by several other large national registries (figure 4), which demonstrate little improvement in survival since the advent of epoprostenol in the early 1990s. This is particularly relevant asi.v.epoprostenol is the only PAH therapy with a demonstrated survival benefit in a single randomised multicentre open-label trial conducted over 30 years ago [16]. There was low use of epoprostenol in initial regimens in our cohort. However, ∼4% of patients in our study were receiving parenteral prostanoids by 2 years in all three time periods, which is slightly lower but still comparable to rates of prostanoid use in other national registries [17,18]. While combination therapy provides unquestionable clinical benefits to patients as demonstrated in several randomised controlled trials [15,19,20], the ultimate impact of sequential or upfront combination therapy on long-term survival remains to be established. To date, trials have historically used a combined clinical morbidity/mortality end-point including a change in 6-min walk distance, which yields equivocal prognostic utility [21–23]. Meta-analyses on these trials have consistently demonstrated an overall benefit of combination therapy on time to clinical worsening, but have produced conflicting reports on mortality [19,20]. At present it is unclear why survival has not improved at our Canadian PAH centres, but our data do not preclude the possibility of improvements in cause-specific mortality from PAH. It is also possible that the lack of improvement was due to <50% of patients being placed on upfront combination therapy after 2015, but by the 2-year follow-up 57% of these patients were on dual or even triple therapy. Thus, another interpretation is that sequential combination therapy strategies have similar benefits to upfront combination therapy with respect to long-term survival. The relatively low rate of initial combination therapy use may be potentially explained by the barriers and delays in access to reimbursement for combination therapy that were present at many Canadian PAH centres until more recent years.
Sequential management with dual or triple therapy is now the reality for many patients who have an inadequate initial response to a single agent or eventual progression to right ventricular failure. The AMBITION trial provided impetus for more aggressive upfront management with combination therapy that is now incorporated into clinical guidelines (table 1).AMBITION investigated the effect of upfront combination therapy with ambrisentan and tadalafil in treatment-naïve patients compared with those receiving monotherapy on the composite end-point of death, hospitalisation, disease progression or unsatisfactory long-term clinical response [15]. Patients placed on upfront dual therapy had a 50% decrease in clinical failure events. Importantly, the primary end-point was largely driven by a decrease in hospitalisation for PAH in patients on combination therapy. In apost hocanalysis of the AMBITION trial data there was a suggestion that survival might be improved for patients who were receiving initial dual therapy, but this requires confirmation [24]. In our study, there was also no difference in survival between patients placed on initial monotherapyversusdual therapy, even after adjusting for baseline mortality risk and disease severity. However, our results do corroborate recent evidence from the French Pulmonary Hypertension Registry that found no difference in long-term survival between those treated with upfront monotherapy or dual therapy in the overall cohort, although there was a small benefit with dual therapy in the subgroup who were intermediate risk at baseline [25]. It is also noteworthy that many modern PAH randomised controlled trials are biased towards selecting a more homogenous population of clinically stable patients without comorbidities. When we applied the AMBITION inclusion/exclusion criteria to the available data in our Canadian cohort, 57% were excluded. Excluded patients were more likely to present with comorbid conditions that conferred a worse prognosis than patients satisfying the inclusion/exclusion criteria for AMBITION.
This Canadian cohort includes patients who were evaluated over an extended period of time during which the management and treatment of PAH has changed. This enabled us to examine whether the evolution of treatment, as reflected in the changing guidelines, has translated to better long-term survival in Canada. The present analysis encompasses PAH patients from three major centres in Canada and represents the largest published Canadian PAH cohort to date. However, there were a few limitations of this analysis. We recognise the limitations in retrospectively collecting data on patients between 2009 and 2017; however, the comparable survival in this group to other international registries argues against a major selection bias. In the present study, we collected data on initial upfront therapy (within 3 months after diagnosis). After the publication of the 2015 ESC/ERS guidelines, the overall number of patients treated with initial combination therapy was still low, relative to their ESC/ERS risk assessment. While some patients after 2015 were only initiated on monotherapy, by the 2-year follow-up 57% of these patients were escalated to dual or even triple therapy, in accordance with their perceived risk or clinical deterioration. Thus, while the use of initial combination therapy was lower than expected, sequential combination therapy was frequently used. We also acknowledge that the propensity score model we used does not account for these later sequential therapy decisions nor does it fully eliminate the possibility of residual confounding, which can only be reduced by randomisation of treatment strategy allocation. The secular trends in PAH survival in the PAH registries also need to be interpreted in the context of changing patient demographics. This is particularly relevant for comparisons with the NIH cohort, which had a mean age at diagnosis that was markedly younger than in more contemporary cohorts [3].
总之,我们的研究的事件PAH患者Canada found that the evolution to a more aggressive initial treatment approach was not associated with incremental improvements in survival in recent years. This study supports the urgent need for new therapies that directly and selectively target disease mechanisms. Other factors such as earlier diagnosis of PAH and personalisation of existing drugs in different combinations continue to be important areas of investigation.
Supplementary material
Supplementary Material
Please note:supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary figuresERJ-01552-2021.Figures
Supplementary tableERJ-01552-2021.Table
Shareable PDF
Supplementary Material
This one-page PDF can be shared freely online.
Shareable PDFERJ-01552-2021.Shareable
Footnotes
This article has an editorial commentary:https://doi.org/10.1183/13993003.00390-2022
Conflict of interest: J.G.E. Zelt has nothing to disclose.
Conflict of interest: J. Sugarman has nothing to disclose.
Conflict of interest: J. Weatherald reports grants, personal fees and nonfinancial support from Janssen Inc. and Actelion, personal fees and nonfinancial support from Bayer, personal fees from Novartis, outside the submitted work.
Conflict of interest: A.C.R. Partridge has nothing to disclose.
Conflict of interest: J. Liang has nothing to disclose.
Conflict of interest: J. Swiston reports personal fees from Janssen J&J, outside the submitted work.
Conflict of interest: N. Brunner reports personal fees from Janssen Pharmaceuticals, outside the submitted work.
Conflict of interest: G. Chandy reports personal fees for advisory board work and consultancy from Bayer Pharmaceuticals, personal fees for advisory board work, lectures and consultancy from Janssen Pharmaceuticals, personal fees for advisory board work from Apotex, during the conduct of the study.
利益冲突:D.J.斯图尔特报告(equity) from Northern Therapeutics, outside the submitted work.
Conflict of interest: V. Contreras-Dominguez has nothing to disclose.
Conflict of interest: M. Thakrar reports grants from Reata Pharmaceuticals, outside the submitted work; and advisory board work for Janssen Pharmaceuticals.
Conflict of interest: D. Helmersen reports grants from Gilead, Janssen and United Therapeutics, outside the submitted work.
Conflict of interest: R. Varughese has nothing to disclose.
Conflict of interest: N. Hirani reports grants (paid to institution) from Bayer, United Therapeutics and Northern Therapeutics, grants (paid to institution) and other (advisory board membership) from Actelion/Janssen, outside the submitted work.
Conflict of interest: F. Umar has nothing to disclose.
Conflict of interest: R. Dunne has nothing to disclose.
Conflict of interest: C. Doyle-Cox has nothing to disclose.
Conflict of interest: J. Foxall has nothing to disclose.
Conflict of interest: L. Mielniczuk reports grants and personal fees for lectures from Janssen, grants from Bayer, outside the submitted work.
Support statement: Funding for this project was provided by the Heart and Stroke Foundation of Canada (G-17-0018315) and the Lung Association of Alberta and Northwest Territories (2019-2020 NGR). Funding information for this article has been deposited with theCrossref Funder Registry.
- ReceivedJune 2, 2021.
- AcceptedOctober 6, 2021.
- Copyright ©The authors 2022.
This version is distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0. For commercial reproduction rights and permissions contactpermissions{at}ersnet.org
References










