





文摘
背景肺泡上皮细胞功能障碍的发病机制中发挥着重要作用特发性肺纤维化(IPF),但仍不完全清楚。肺纤维化的一些单基因形式与表达相关突变体表面活性剂蛋白C (SFTPC)。最常见的致病突变,I73T mislocalises肺泡上皮细胞的质膜和显示一个有毒增益函数。因为解释这个突变和IPF之间的联系机制不完全理解,我们试图询问SFTPC贩卖理解健康和疾病的功能性意义SFTPC-I73T relocalisation。
方法我们进行机械的分析SFTPC贩卖,繁殖的细胞模型在活的有机体内在人类原始肺泡瀑样表型和验证结果。
结果我们表明,野生型SFTPC意外间接走私路线通过等离子体膜和经历多个卵裂的第一个事件之前多泡体(多功能车辆总线)进行进一步处理。SFTPC-I73T需要同样的路线,但其进展延迟在细胞表面和由于贩卖到多功能车辆总线的失败。无法接受开始贩卖,它是质膜回收部分中间分开。
结论这些数据显示,所有SFTPC首次通过细胞表面在正常交易,和I73T突变积累在细胞表面通过推迟贩运和活跃的回收。这种理解正常的SFTPC贩运和I73T突变如何扰乱它提供了新颖的见解SFTPC生物学在健康和疾病,并在IPF发展SFTPC变异的贡献。
文摘
研究SFTPC突变导致家族性肺纤维化可能帮助我们开发IPF的新疗法。通过研究致病突变,本研究揭示了新见解SFTPC处理在肺泡细胞,这是如何摄动的疾病。https://bit.ly/344VUhm
介绍
在特发性肺纤维化(IPF),病理性瘢痕的肺损害气体交换导致过早死亡(1]。2型(at₂)肺泡上皮细胞,细胞合成表面活性剂和AT1细胞的祖细胞(2),在IPF发展是至关重要的。越来越认可的是,at₂在IPF触发病理损伤和功能障碍方面研究肺改造(3),但有限的理解在IPF初始的事件阻碍发展的早期治疗,直接修改致病机制。基维辛迪家族性肺纤维化),由单个基因的突变引起的,提供了一个独特的机会来审视特定at₂缺陷导致肺纤维化(4]。
在表面活性剂常染色体显性突变蛋白C (SFTPC),这是表示只在at₂细胞,导致基维辛迪[5]。最常见的致病变种,SFTPC-I73T [6),可以表现为基维辛迪在儿童或成人(6,7]。其中一些变异SFTPC不流量超出内质网(ER),因为他们无法正确折叠。这导致“ER应激”(8),这是在肺部基维辛迪和零星的IPF (9]。然而,SFTPC-I73T不会导致ER压力,而是一直报道mislocalise细胞表面的未知的机制(10]。
理解SFTPC贩运是必要的,如果我们要解释FPF-causing SFTPC-I73T变异导致蛋白质mislocalisation和at₂功能障碍。SFTPC proprotein生产(proSFTPC),经历了几个分泌蛋白水解步骤之前,尽管令人惊讶的是成熟的步骤的精确位置和性质尚不清楚。ProSFTPC 2型跨膜蛋白,包括四个域名(图1一个):1)一个氨基端胞质域post-Golgi所需目标(11- - - - - -14];2)跨膜螺旋形成成熟的SFTPC蛋白(15];3)非结构化连接器领域;和4)c端BRICHOS域确保正确的蛋白质折叠。当前模型表明proSFTPC交通量直接从早期细胞分泌途径的隔间专业晚期核内体称为多泡体(多功能车辆总线),然后AT2-specific片状体分泌(之前16]。途中,连续的C - n端分裂产生成熟SFTPC (图1一个)[17]。然而,I73T突变干扰这个过程导致异常积累成熟SFTPC月初在质膜和核内体(10,18]。据报道,这是毒性at₂功能,干扰磷脂和蛋白质降解吸收通过自噬和mitophagy18,19]。了解I73T突变阻止正常贩卖SFTPC可能最终使治疗的发展,防止at₂损伤和功能障碍,并防止纤维化的发病。
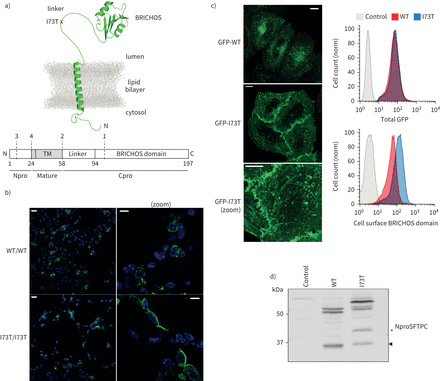
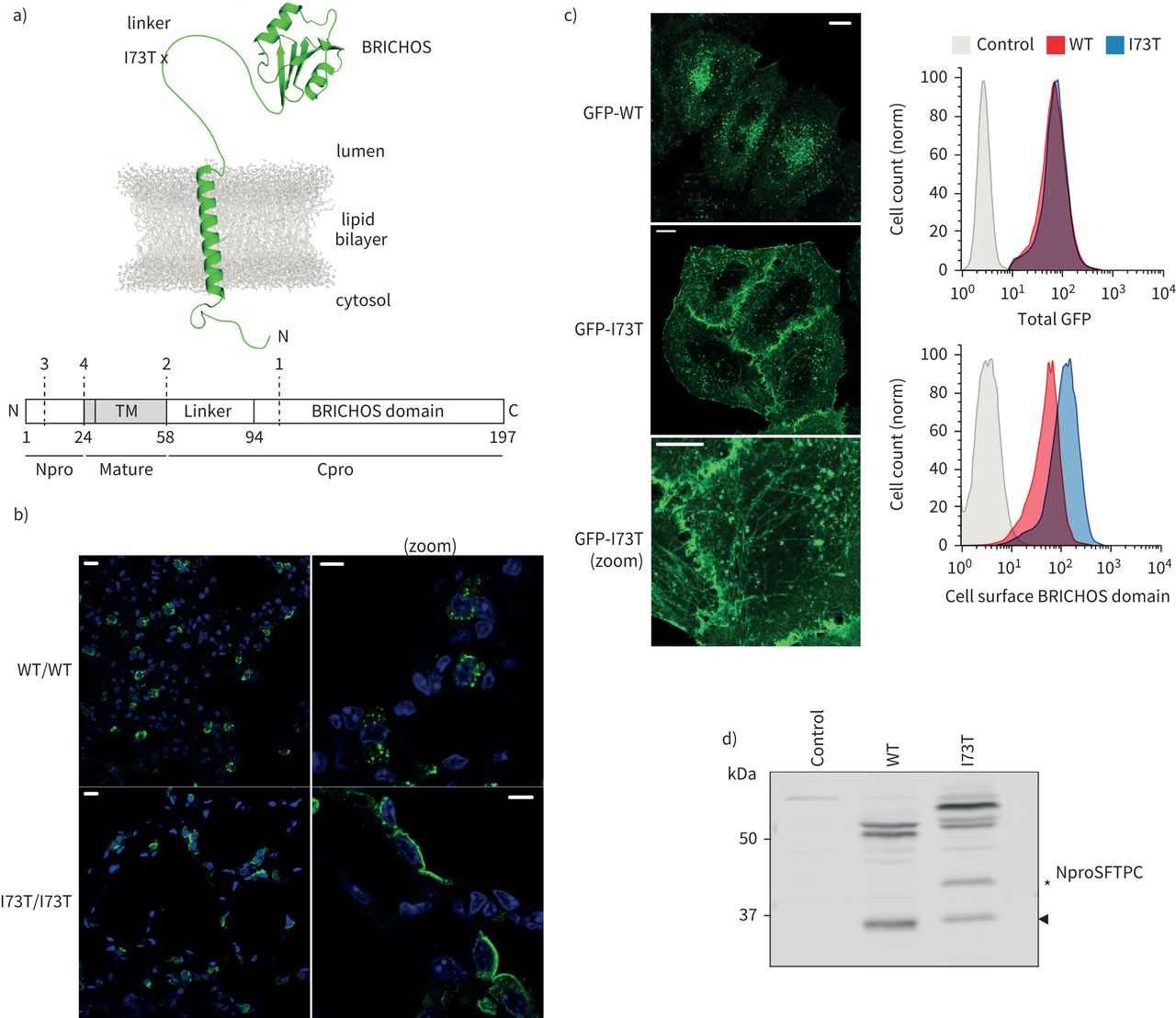
表面活性剂蛋白C (SFTPC) -I73T异常和处理在活的有机体内和在体外。SFTPC)图表的描述结构和致病性I73T突变的位置以及域和近似乳沟网站报道(虚线,编号1 - 4)。b) ProSFTPC从转基因小鼠肺泡组织化学染色组织有条件地表达SFTPC-WT(野生型)或SFTPC-I73T揭示点状的细胞内染色SFTPC-WT和再分配SFTPC-I73T顶端质膜。c)表达绿色荧光蛋白(GFP) -SFTPC-WT时在海拉细胞定位在胞内点状的结构,而GFP-SFTPC-I73T relocalises质膜和细胞内的管状结构。细胞中表达等量的GFP面板(右上),有过多的细胞表面SFTPC-I73T以BRICHOS域抗体(右下面板)。d)免疫印迹的溶解产物从矢量控制或使用NproSFTPC GFP-SFTPC表达细胞抗体显示改变SFTPC-I73T的处理。TM:跨膜。规模酒吧= 10μm或5μm(放大图片)。
在这里,我们提出一个详细的分析SFTPC贩卖首次显示,野生型和I73T-mutant SFTPC交通意外通过等离子体膜。Mislocalisation SFTPC-I73T结果异常的回收的蛋白质细胞表面由于其未能针对多功能车辆总线,导致其积累在细胞表面和回收核内体。我们的调查的一种罕见的致病性变异从而提供基本见解SFTPC的生物学和疾病的机制。
材料和方法
详细的方法包括化学物质的细节,抗体细胞株,小干扰rna (si), CRISPR指南,建立表达载体和细胞化验是可用的补充材料。
细胞培养
海拉细胞被维护在DMEM + 10%胎牛血清(+ 400µg·毫升−1geneticin稳定细胞株)。DNA转染是通过使用FuGene 6 (Promega)和克隆转基因细胞系生成的选择和单细胞排序。淘汰赛细胞池通过脂质体转染生成向量表达Cas9,引导RNA,荧光标记用于fluorescence-activated细胞排序。
瀑样文化
肺泡瀑样得到使用feeder-free organoid-based扩张tissue-isolated主要肺泡细胞的方法。手稿描述该方法在制备的细节(个人通信,桑德van Riet,莱顿大学医学中心,莱顿,荷兰)。短暂,发表周围肺组织收集病人接受癌症切除并受酶消化。使用收集的肺组织的框架内病人护理符合荷兰医学科学学会联合会的行为准则(20.]。本行为准则描述了退出系统编码匿名进一步使用这样的样本。细胞培养中瀑样基底膜提取瀑样矩阵使用feeder-free条件。瀑样保留at₂阳性染色细胞特征如图所示的proSFTPC和htii - 280和通道1用于这些实验。
结果
与野生型相比SFTPC, mislocalised I73T变体,misprocessed O-glycosylated异常
当检测到使用n端结构域(Npro)抗体,我们证实了野生型proSFTPC (SFTPC-WT)本土化点状的结构符合多功能车辆总线(21,22),重新分配SFTPC-I73T变体at₂细胞的质膜SFTPC-I73T转基因小鼠肺(图1 b)[10,18]。
调查机制负责,早期SFTPC贩卖成立的细胞模型。主要at₂细胞不适合详细的机械的工作由于所需基因操作的复杂性和其去分化在单层培养(23]。海拉细胞一直被用来进行研究的蛋白质贩卖,已经存在大量生物转基因海拉行来审问通用生物过程顺行交易负责。增强型绿色荧光protein-proSFTPC (GFP-SFTPC)表达在海拉细胞GFP-tag并不影响本地化而未加标签的蛋白质(补充图S1a)。表面积累相对于GFP-SFTPC-WT GFP-SFTPC-I73T可以通过显微镜和流式细胞术检测使用抗体BRICHOS域,但有趣的是,我们可以检测到低水平的SFTPC-WT质膜(图1 c)。此外,GFP-SFTPC-I73T积累在管状结构(图1 c)MICAL-L1和Rab8 co-staining证明回收核内体(补充图印地)。
SFTPC从细胞溶解产物迁移物种的多个大小没有或GFP标记(图1 d和补充图就是S1c)。GFP-SFTPC-WT迁移∼50 kDa的紧身上衣和一个小乐队∼34 kDa(箭头)。紧身上衣代表长篇proSFTPC±棕榈酰化(15,24]。按照先前的研究[10),SFTPC-I73T迁移更慢,积累额外的物种“SFTPC *”(*,图1 d和补充图就是S1c)。Affinity-purified GFP-SFTPC-WT和GFP-SFTPC-I73T进行质谱分析,证实了上面的乐队是完整的蛋白质,而最小的乐队缺乏BRICHOS域和链接器,和SFTPC *乐队GFP-SFTPC-I73T样本缺乏BRICHOS域,但保留链接器(补充图S1d)。
的相关性来确定I73残渣SFTPC成熟期间,生成GFP-SFTPC-I73A变体。像GFP-SFTPC-I73T,这局部的质膜(补充图S2a)和累积SFTPC *——物种(补充图开通)。然而,乐队的大小类似于GFP-SFTPC-WT GFP-SFTPC-I73A迁移,尽管比例与GFP-SFTPC-I73T相似。O-glycosylation经常发生在苏氨酸残基膜蛋白在贩卖的25),所以我们提出缺陷SFTPC-I73T可能反映O-glycosylation T73。这是证实了质谱分析,通过观察O-glycosidase治疗恢复的迁移GFP-SFTPC-I73T GFP-SFTPC-WT(的补充图S2c和d)。
突变的I73因此导致SFTPC积累在细胞表面和回收核内体,并促进积累的uncleaved proSFTPC和部分加工SFTPC *形式,保留链接器,但缺乏BRICHOS域。这表明I73扮演着一个重要的角色在SFTPC贩运和蛋白质水解的链接器区域。73 t的观察O-glycosylation不负责SFTPC mislocation,但似乎也可以理解为自然的毒性作用。
质膜SFTPC-WT交通量
ProSFTPC被认为交通直接从高尔基体蛋白水解作用的酸性室(26,27]。直接高尔基贩卖核内体、胞质适配器(如。AP1或GGA蛋白质)承认针对主题(如。YXXØ或(DE) XXX (LL))促进包装成clathrin-coated囊泡(28]。胞质域SFTPC缺乏这样的序列,但GGA蛋白质(特别是GGA2)能够识别一些ubiquitinated货物缺少针对序列(29日]。因为SFTPC ubiquitinated赖氨酸6(转K6),可能允许gga识别,我们拆卸GGA1-3,但发现不影响GFP-SFTPC本地化或蛋白水解作用(补充图S3a和b)。然而,由于适配器蛋白质击倒后允许细胞逐渐耗尽的补偿,我们下一个测试的影响迅速GGA2使用“knock-sideways”GGA2失活是relocalised线粒体瞬间在添加雷帕霉素(30.]。保留使用选择性钩子(冲)系统启用可视化SFTPC贩卖活细胞(图2一个)[31日]。Streptavidin-binding蛋白(SBP) -GFP-SFTPC最初局部ER,但是在添加生物素relocalised模式类似GFP-SFTPC (图2 b)。biotin-induced释放后,SBP-GFP-SFTPC-WT贩卖到远端隔间包括质膜,虽然没有影响GGA2-knocksideways观察(补充图S3c和d)。这让我们假设所有SFTPC,包括野生型,可能交通通过细胞表面,而不是直接从高尔基体核内体。
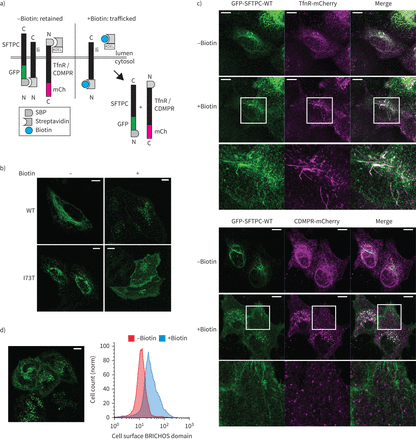
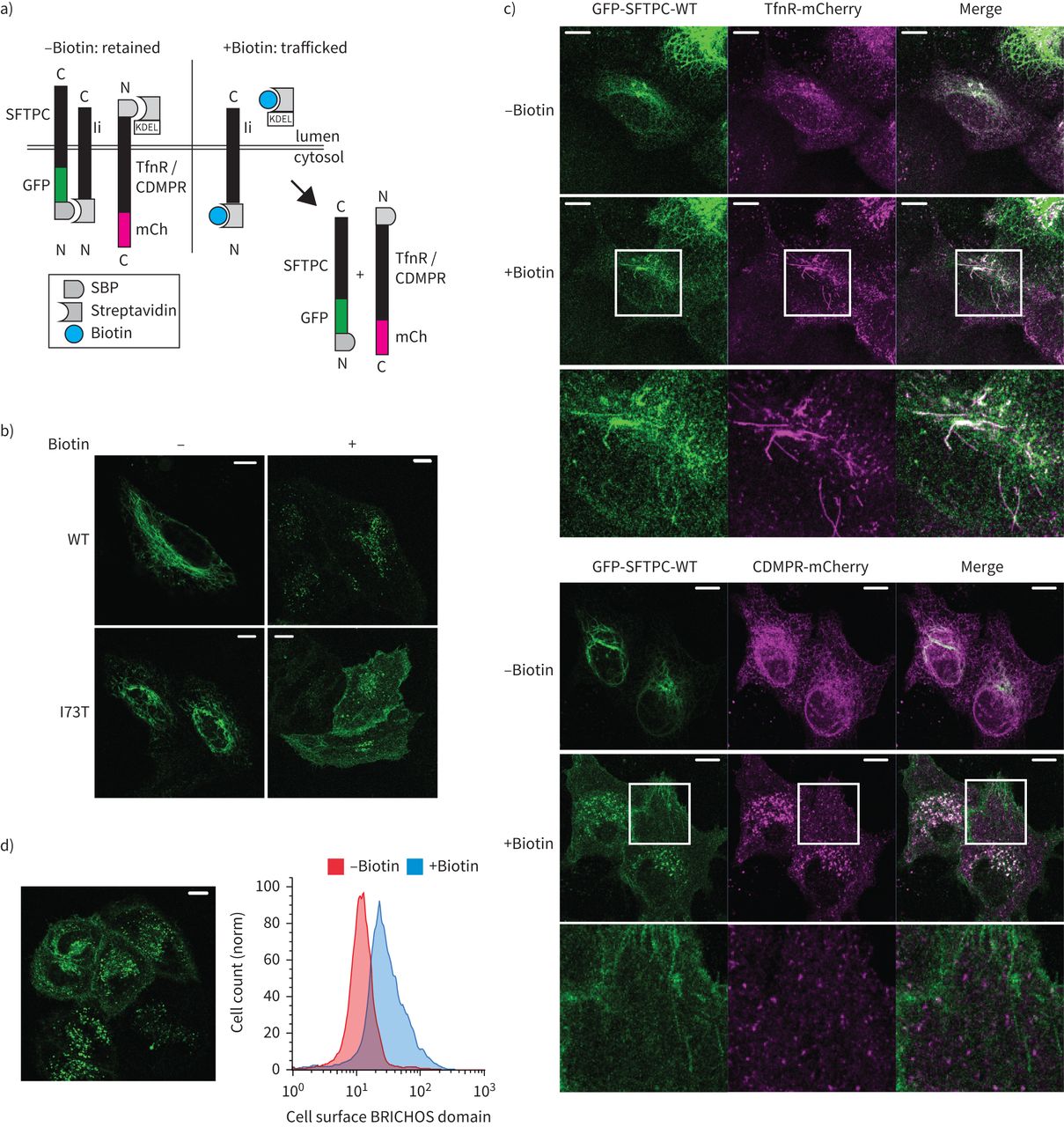
所有表面活性剂蛋白C (SFTPC)跟踪从早期隔间质膜。使用选择性的钩子)保留的示意图(冲)系统,在使用链霉亲和素蛋白在早期跟踪隔间举行“钩”之前释放之后添加生物素。b)本地化SFTPC的海拉细胞表达RUSH-green荧光蛋白(GFP) -SFTPC融合蛋白治疗±生物素16 h证实他们交通通常post-Golgi隔间。c)的实时跟踪与转铁蛋白受体(TfnR)或CDMPR GFP-SFTPC揭示co-localisation SFTPC TfnR在管状结构,但失败与CDMPR co-localisation超出了高尔基的囊泡。d)生物素2 h后曝光,GFP-SFTPC-wild类型(WT)是可见的在质膜;这反映在增加BRICHOS域出现在流式细胞仪的质膜。SBP: streptavidin-binding蛋白质、妇幼保健:mCherry。酒吧= 10μm规模。
为了验证这一点,我们比较SFTPC贩卖与转铁蛋白受体(TfnR)和CDMPR,交通通过等离子体膜或直接endolysosomal系统,分别为(图2一个)[32]。引人注目的是,SFTPC-WT贩卖通过管状与TfnR post-Golgi结构,没有与CDMPR co-localisation,退出了高尔基体小泡(图2 c)。正如所料,SBP-GFP-SFTPC-I73T也贩卖TfnR然后贩卖到质膜(补充图S4a和b)[10]。值得注意的是,SBP-GFP-SFTPC-WT也贩卖到质膜,所呈现的影像和流式细胞术(图2 d)。
这些数据表明,SFTPC-WT和SFTPC-I73T交通直接从高尔基体等离子体膜。因此,SFTPC-I73T的积累在细胞表面代表异常的保留,而不是mistrafficking本身。
检索SFTPC-WT AP2-dependent从质膜内吞作用然后目标起K63 oligo-ubiquitination
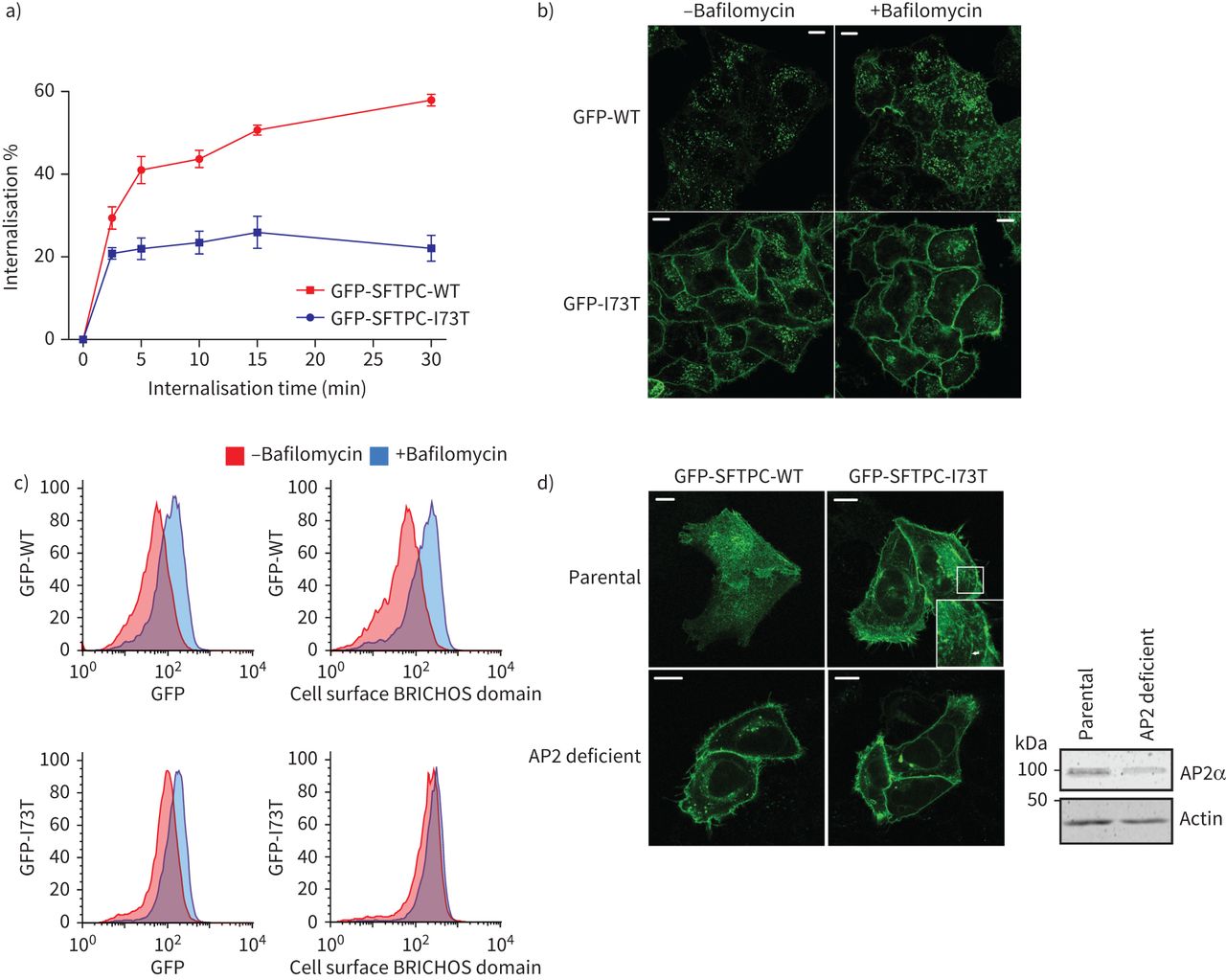
我们接下来研究贩卖SFTPC从质膜使用抗体喂养:细胞表面SFTPC贴上了初级抗体在冰上,在37°C的环境允许掩饰,然后剩下的细胞表面蛋白使用荧光共轭二次抗体的检测。标签GFP-SFTPC-WT因为比GFP-SFTPC-I73T完全主观的,尽管有一些早期SFTPC-I73T掩饰的证据(图3一)。这是荧光GFP标记的影响引入到想象贩卖(补充图S5)。稳定期I73T信号可能反映了回收标签蛋白回到质膜过程中测定。初始SFTPC掩饰的速度符合clathrin-mediated内吞作用(CME)。bafilomycin治疗时间延长,从而抑制芝加哥商品交易所(33),导致积累GFP-SFTPC-WT质膜,但对GFP-SFTPC-I73T的分布影响很小(图3 bc),芝加哥商品交易所确认参与AP2-deficient细胞,GFP-SFTPC-WT和GFP-SFTPC-I73T都累积在质膜(图3 d)。
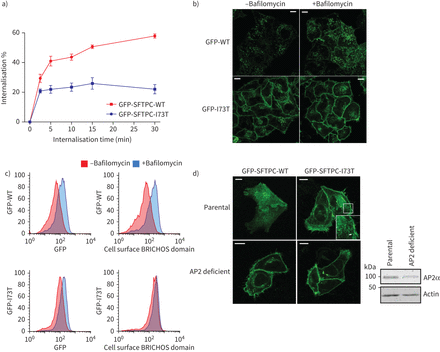
表面活性剂从质膜蛋白C (SFTPC)掩饰I73T AP2依赖,弱智的突变。一)定量SFTPC掩饰化验。细胞表达绿色荧光蛋白(GFP) -SFTPC-wild类型(WT)或-I73T与BRICHOS域标记抗体在冰上,那么蛋白质允许员工在37°C表示。细胞被放置在冰,固定但不permeabilised,并贴上了一个二级抗体之前通过流式细胞术分析。n = 3。数据意味着±扫描电镜。b)和c)海拉细胞稳定表达GFP-SFTPC-WT或-I73T服用30µM bafilomycin 16 h抑制clathrin-mediated内吞作用。这导致细胞表面SFTPC积累所看到的共焦显微镜和流式细胞仪测量。d) AP2-deficient细胞被用来评估本地化使用选择性保留的钩子(冲)ed GFP-SFTPC-WT和-I73T 2 h后生物素治疗。AP2α-subunit AP2M1 CRISPR淘汰赛细胞免疫印迹。尽管一些α-subunit依然存在,这个复杂的非功能。酒吧= 10μm规模。
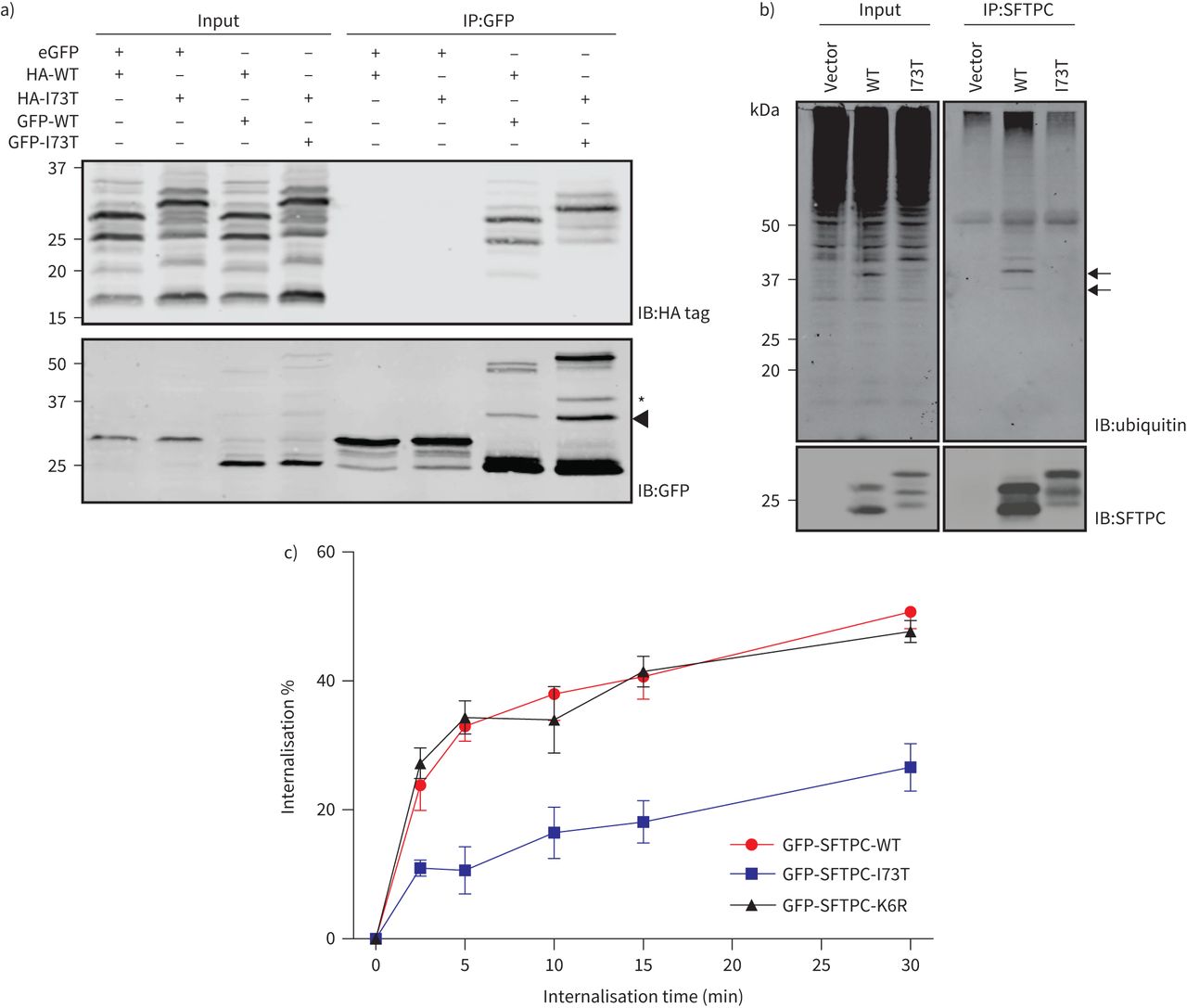
因此,I73T突变部分损害CME尽管拓扑无法与排序AP2等因素。许多膜蛋白进行集群促进芝加哥商品交易所。的确,oligomerisation SFTPC是发生在其交易(21]。然而,免疫沉淀反应中的GFP-SFTPC和HA-SFTPC产生hetero-oligomers SFTPC-WT和SFTPC-I73T,表明受损的内吞作用SFTPC-I73T oligomerisation不太可能用失败来解释(图4一)。
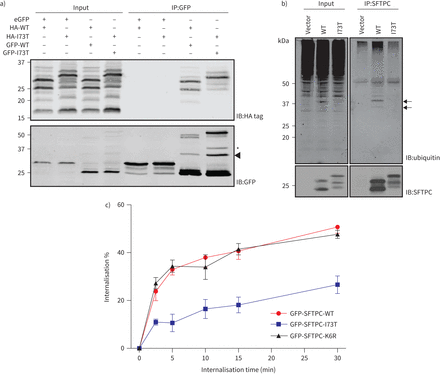
缺陷表面活性剂蛋白C (SFTPC) -I73T掩饰失败并不是因为oligomerisation或泛素化。一)海拉细胞转染与血凝素(HA)标记和绿色荧光蛋白(GFP)标记SFTPC(野生型(WT)或I73T)和溶解产物受到anti-GFP免疫沉淀反应之前HA标记和绿色荧光蛋白免疫印迹。SFTPC-WT和SFTPC-I73T都同样能够oligomerise。b)从海拉细胞溶解产物表达SFTPC-WT或SFTPC-I73T受到SFTPC免疫沉淀反应和泛素的免疫印迹。可见oligoubiquitinated SFTPC-WT(箭头),这是在I73T-expressing细胞未见。c)定量SFTPC掩饰测定细胞内表达GFP-SFTPC-WT I73T或K6R。n = 3。数据意味着±扫描电镜。
我们下一个被认为是异常的泛素化的可能性SFTPC-I73T导致交易中断。SFTPC ubiquitinated转K6残留,据报道已参与endosomal目标(13,14]。免疫沉淀反应的无标记和GFP-tagged SFTPC证实oligoubiquitination缺席的SFTPC-WT SFTPC-I73T (图4 b和补充图S6a)。我们生成的GFP-SFTPC-K6R和证实这不是ubiquitinated (补充图S6b)。然而,SFTPC-K6R掩饰SFTPC-WT表明泛素化的区别是不必要的内吞作用,因此不能占之间的内吞作用的不同利率SFTPC-WT和SFTPC-I73T (图4 c和补充图S5)。
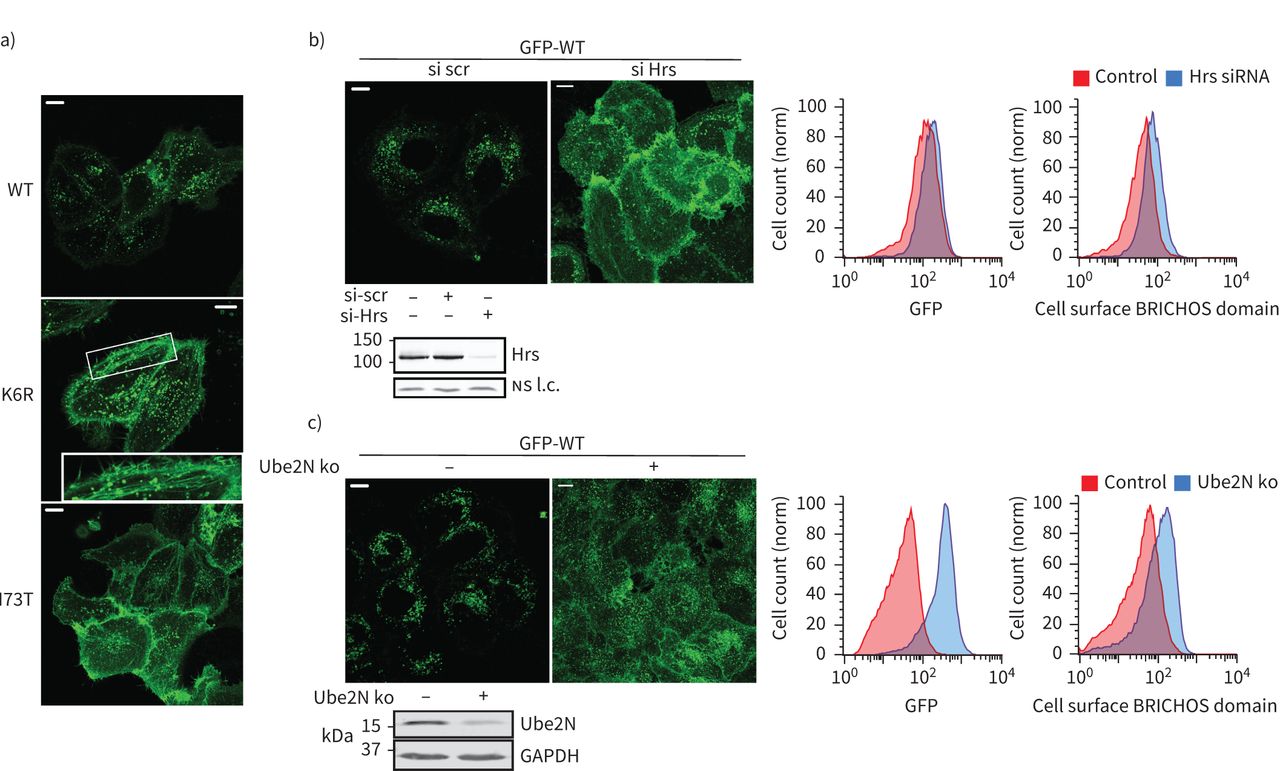
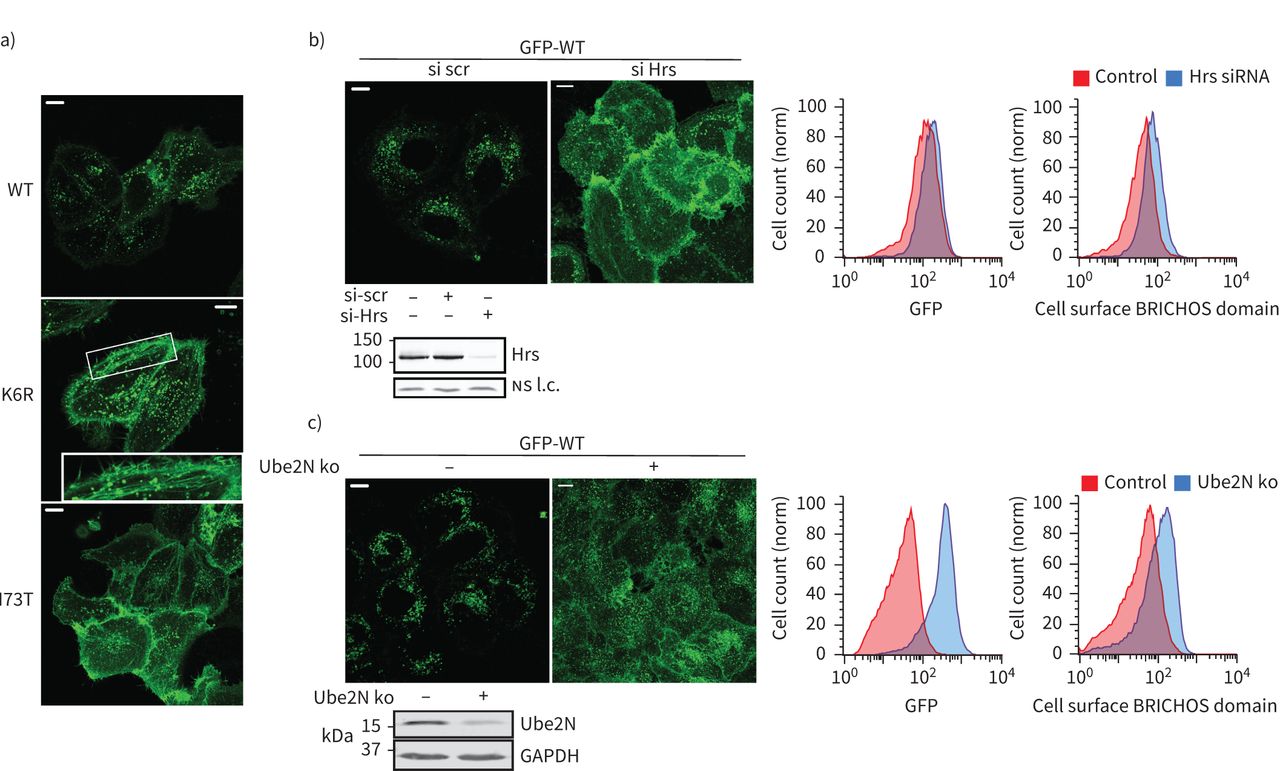
回收的观察积累SFTPC-I73T核内体显示排序在核内体向多功能车辆总线的失败。通过显微镜,我们观察到ubiquitination-deficient GFP-SFTPC-K6R局部的质膜和回收核内体(图5一个和补充图S6c)。自掩饰的蛋白质进入管腔内的囊泡的多功能车辆总线涉及识别endosomal-sorting ubiquitinated货物的运输所需的复杂(ESCRT)机械34],我们耗尽细胞ESCRT0蛋白质小时[35)和观察到的一个戏剧性的relocalisation GFP-SFTPC-WT回收核内体和质膜(图5 b)。K63 E2-ligase产生的泛素链(Ube2N) [36]马克对贩卖蛋白质核内体34]。Ube2N耗尽时,GFP-SFTPC-WT再次重新回收核内体和质膜(图5 c)。
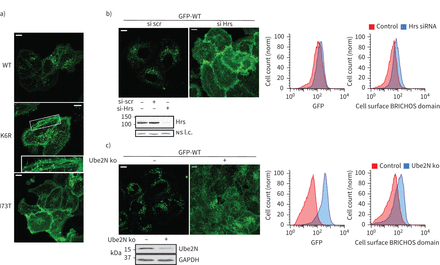
表面活性剂蛋白C (SFTPC)泛素化K63链需要追踪到多泡体(多功能车辆总线)。)共焦成像的海拉细胞表达绿色荧光蛋白(GFP) -SFTPC-wild类型(WT) ubiquitination-deficient K6R I73T表明,像I73T, SFTPC-K6R重新分配到细胞表面,进入回收核内体(中间面板中,插图)。b)海拉细胞转染炒(scr)或ESCRT0蛋白质小时siRNA 48 h。免疫印迹证实击倒和共焦成像显示的再分配GFP-SFTPC-WT质膜。流式细胞术证实略有增加总GFP +部分重新分配SFTPC质膜的小时核的存在。c) Ube2N CRISPR淘汰赛池和成功经免疫印迹。共焦显微镜显示两个GFP信号的整体提升和再分配GFP-SFTPC-WT这些细胞的质膜,经流式细胞术。ns:不重要的;l.c。:loading control. Scale bars=10 μm.
这些结果说明SFTPC-WT和SFTPC-I73T内源性AP2-dependent方式,尽管SFTPC-I73T内化效率较低。K63 SFTPC-WT然后指导它的泛素化多功能车辆总线管腔内的囊泡。这个排序事件的失败重定向SFTPC-I73T质膜通过回收核内体。
之前SFTPC经历c端乳沟内吞作用后进入多功能车辆总线
蛋白水解的成熟SFTPC发生在酸性post-Golgi隔间(26,37]。一个组织蛋白酶H-mediated n端乳沟在多功能车辆总线27之前是c端分裂在一个未知的位置。乳沟事件发生之前多功能车辆总线条目可以在海拉细胞研究,而评估的氨基端蛋白质水解后车厢需要的所有SFTPC贩卖隔间(包括层状体),因此没有进一步研究。
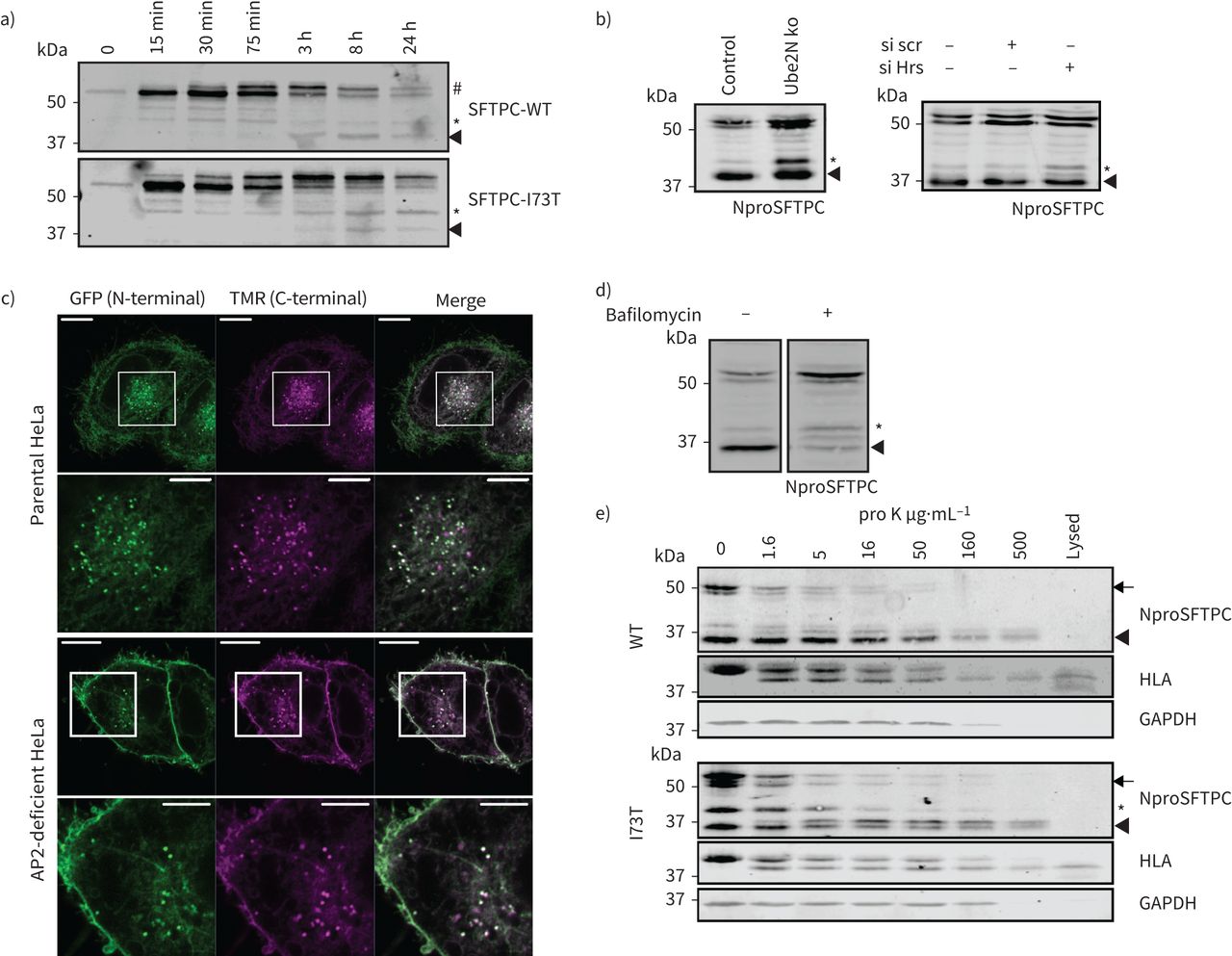
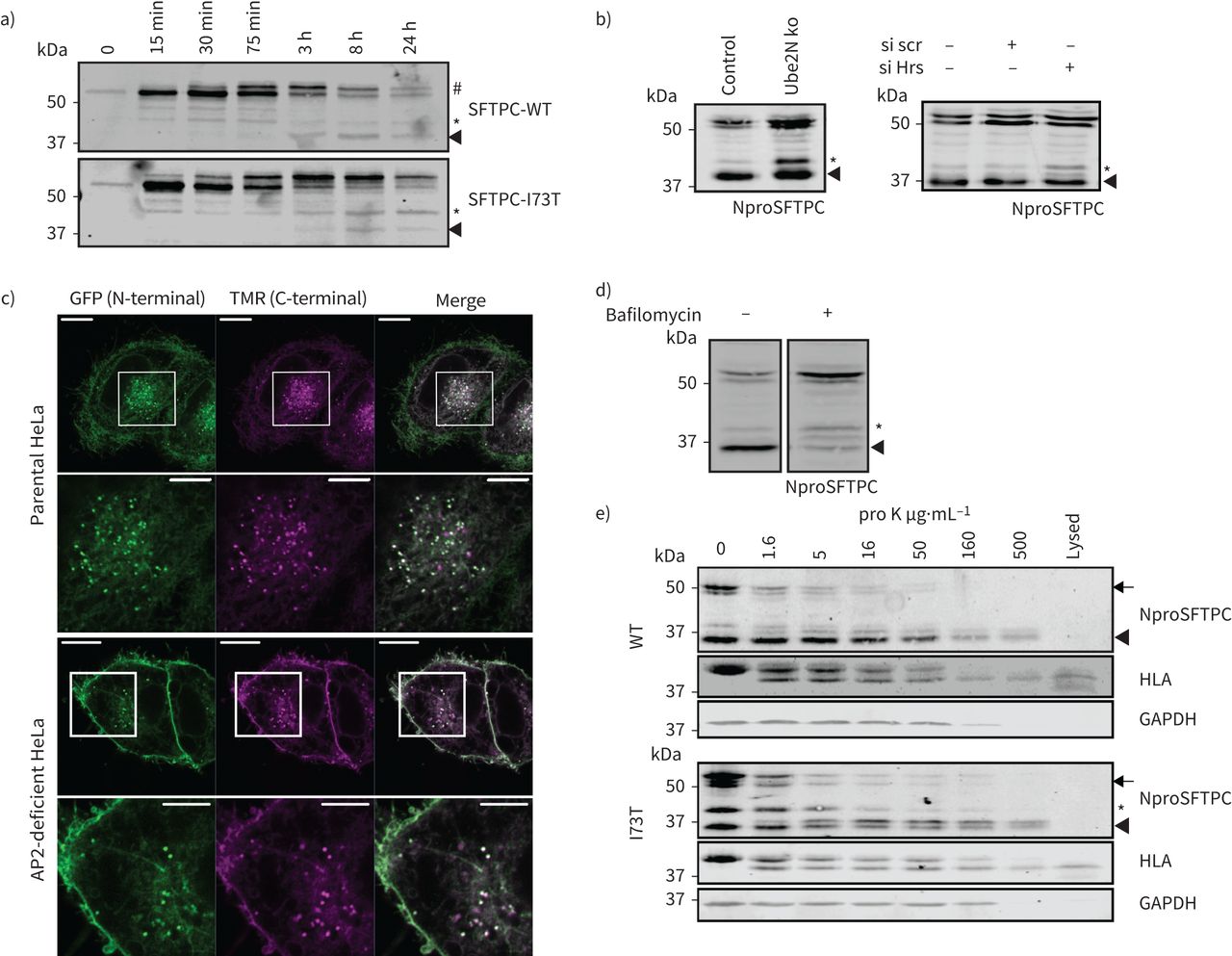
使用系统,在30分钟后释放我们观察到全身SFTPC-WT和SFTPC-I73T都经历了棕榈酰化导致较慢的迁移带视觉效果的免疫印迹(#;图6)。从3 h,全身SFTPC-WT丢失而37-kDa c端裂解产品出现(箭头)。相比之下,保留更多的长篇SFTPC-I73T和SFTPC *产品积累。类似的乐队在SFTPC-WT-expressing早期观察细胞,但消失3 h。我们得出结论,早期c端蛋白质水解生成SFTPC *发生在一个隔间SFTPC-WT和SFTPC-I73T访问。选择性积累SFTPC * SFTPC-I73T表达式表明,连续的c端处理由I73T受损。确定这是一个原因或异常交易的结果,我们删除Ube2N或耗尽ESCRT0蛋白质小时损害多功能车辆总线导入。都引起WT-SFTPC *积累,这表明访问为最后c端劈理(多功能车辆总线是必要的图6 b)。持续的低水平的c端裂解SFTPC在所有条件,表明一个不完整的贩卖块或一些冗余SFTPC排序。
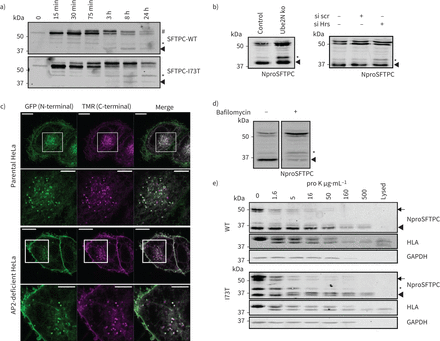
表面活性剂蛋白C (SFTPC) C端乳沟过境后发生通过质膜,但之前多泡体(多功能车辆总线)内在化。)海拉细胞表达保留使用选择性钩子(冲)-SFTPC融合蛋白被允许流量前《纽约时报》表示溶解产物受到绿色荧光蛋白(GFP)免疫沉淀反应和NproSFTPC免疫印迹。中间形式(*和箭头)出现同时SFTPC-wild类型(WT)和-I73T,但是第一I73T中间(*)积累WT中间被清除。b)海拉细胞缺乏Ube2N或小时SFTPC不能进入多功能车辆总线积累SFTPC *。c)海拉细胞转染与GFP-SFTPC-Halo Halotag标签与咯配体修复前15分钟。d)治疗GFP-SFTPC-WT表达海拉细胞与100 nM bafilomycin 16 h导致优惠长篇proprotein积累。e)海拉细胞稳定表达GFP-SFTPC-WT或I73T孵化与蛋白酶K消化暴露在等离子体膜蛋白是细胞溶解和SFTPC受到免疫印迹,人类白细胞抗原(HLA)和GAPDH。全身(WT和I73T)和中间(SFTPC *)物种被蛋白酶K消化,建议他们居住至少部分在细胞表面。细胞溶解,溶解在蛋白酶K治疗。规模酒吧= 10μm或5μm(放大图片)。
确定c端蛋白水解作用的亚细胞位置,我们生成的GFP-SFTPC-WT-HaloTag和确认其正确的交易和乳沟(补充图S7)。大多数GFP-SFTPC-WT-HaloTag-containing puncta保留这两个标签,确定他们是车厢前c端蛋白水解作用(图6 c)。在AP2-deficient细胞,uncleaved GFP-SFTPC-WT-HaloTag积累在质膜,表明全身SFTPC首先到达细胞表面和内吞作用后发生c端蛋白水解作用。这一发现进一步支持的观察,bafilomycin-mediated抑制内吞作用导致积累完整proSFTPC但有一个相对较小的影响*中间(丰度图6 d)。
我们试图确定c端受损的蛋白水解作用有助于mislocalisation SFTPC-I73T或结果。流仪检测细胞表面BRICHOS域(uncleaved蛋白质)显示SFTPC-WT不那么引人注目的差异和SFTPC-I73T比成像观察到胞质GFP-tag (图1 c)。当SFTPC-WT已经得到了相似的结果重新分配损害的细胞表面endosomal排序(图5 b和c)。这表明回收SFTPC回收之前可能接受c端蛋白水解作用。调查,我们对完整细胞蛋白酶K消化surface-exposed蛋白(图6 e)。人类白细胞抗原(HLA)作为控制表面接触,虽然GAPDH胞质蛋白水解作用。全身SFTPC-WT和SFTPC-I73T被低浓度的蛋白酶K消化HLA消化所需类似,确认他们的存在在细胞表面。相比之下,完全c端裂解37-kDa乐队拒绝蛋白质水解程度类似于GAPDH暗示一个细胞内的本地化。SFTPC *显示一个中间对蛋白酶K,符合混合表面和endosomal本地化。随着蛋白酶K消化了冰,没有贩卖SFTPC可能发生在这个孵化。因此,失去SFTPC *中间反映其蛋白酶K消化,而不是失去全身proSFTPC的间接影响。
全身SFTPC因此首先贩卖到细胞表面,然后后AP2-dependent内吞作用,c端蛋白质水解发生远端连接器领域产生SFTPC *。这是紧随其后的是juxta-membrane解理完全生成c端SFTPC分开。虽然SFTPC-WT然后迅速运往氨基端蛋白水解作用的多功能车辆总线,I73T突变损害juxta-membrane乳沟,导致回收SFTPC *到细胞表面。
SFTPC乳沟是必要的目标从细胞表面多功能车辆总线
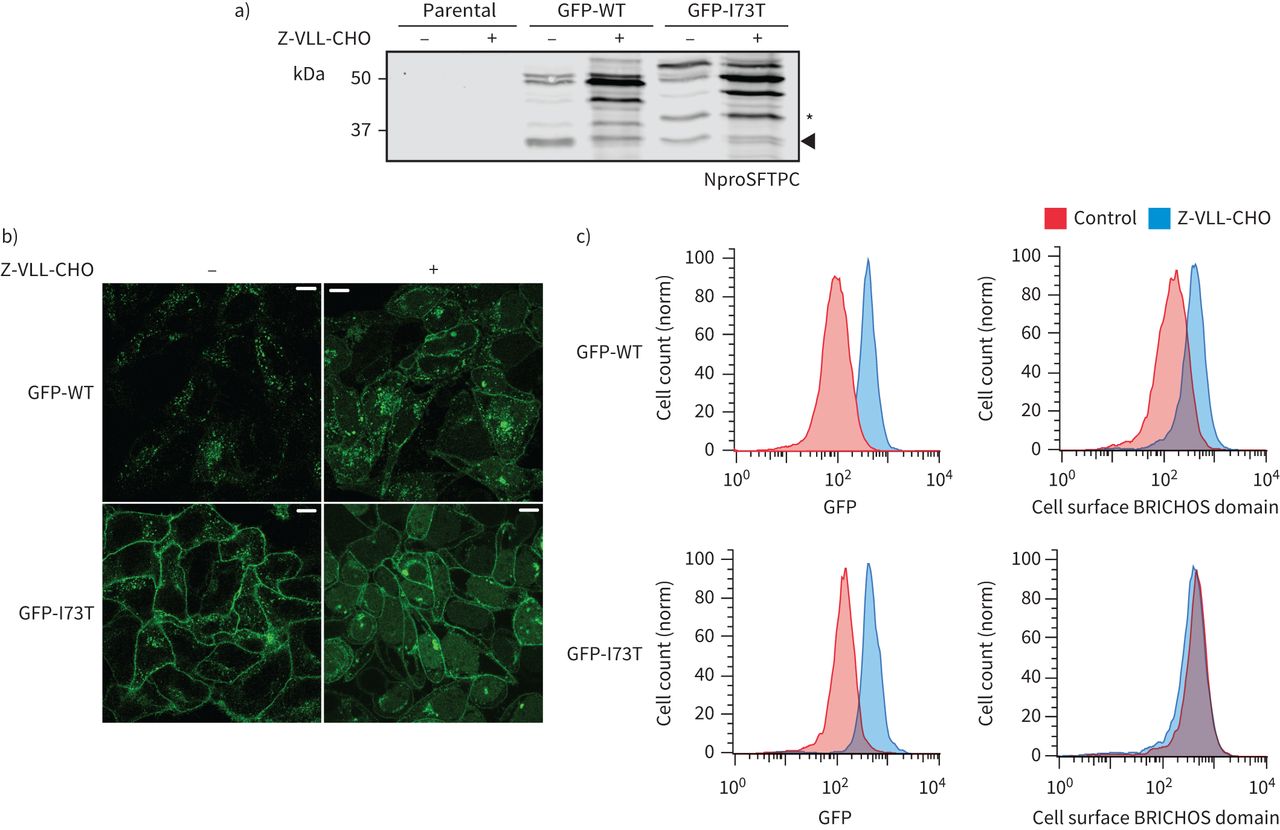
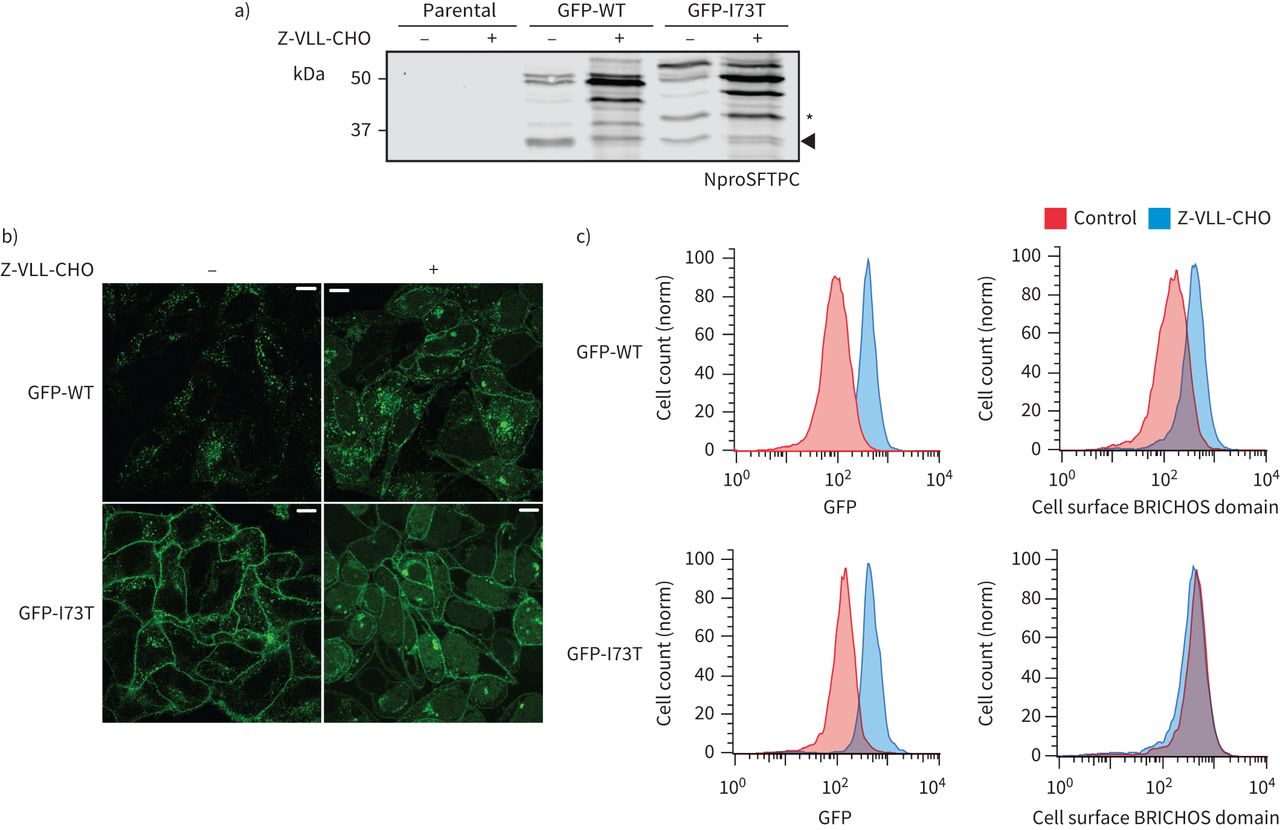
我们希望确定蛋白水解切除链接器区域作为一个信号转移SFTPC多功能车辆总线和回收到质膜。另一个BRICHOS-domain-containing蛋白质,Bri2,经历了连续的劈理首先furin远离膜,然后靠近细胞膜的膜结合sheddase ADAM10 [38]。我们试图插入标记之间的跨膜域和链接器受损juxta-membrane乳沟尤其是长标签(补充图S8)。这不成比例地影响SFTPC-WT导致积累SFTPC *式的产品。怀疑juxta-membrane乳沟可能由一个endosomal膜结合sheddase,我们与Z-VLL-CHO细胞治疗,这样的蛋白酶抑制剂。Z-VLL-CHO SFTPC-WT水平大幅度增加溶解产物,主要为全身蛋白质和BRICHOS-cleaved中间体(图7)与SFTPC-WT积累在细胞表面(图7 b和c)。蛋白水解处理SFTPC-I73T被Z-VLL-CHO进一步受损,和SFTPC-I73T仍然在细胞表面。
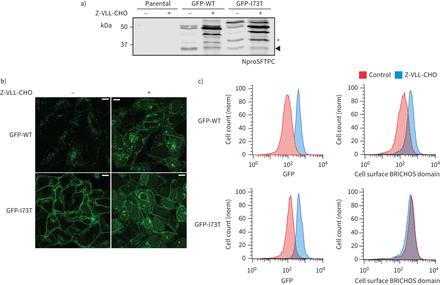
表面活性剂蛋白C (SFTPC) C端乳沟被Z-VLL-CHO抑制。一)海拉细胞稳定表达绿色荧光蛋白(GFP) -SFTPC-wild类型(WT)或-I73T服用5μM Z-VLL-CHO 16 h和溶菌产物NproSFTPC免疫印迹。细胞治疗Z-VLL-CHO开发过度的早期c端处理中间体,增加整体SFTPC(以GFP)和再分配SFTPC质膜,在b)共焦显微镜和流式细胞术。酒吧= 10μm规模。
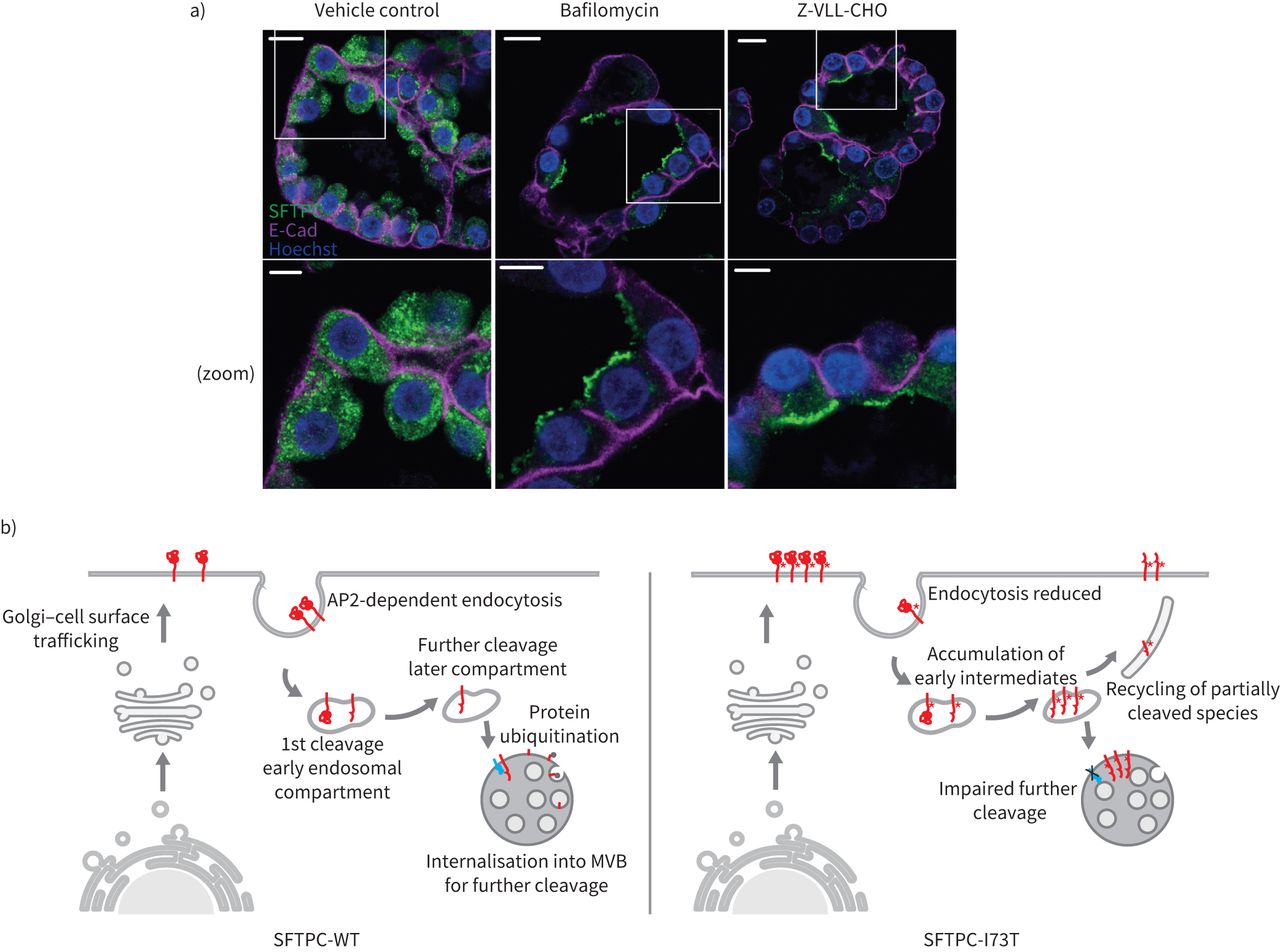
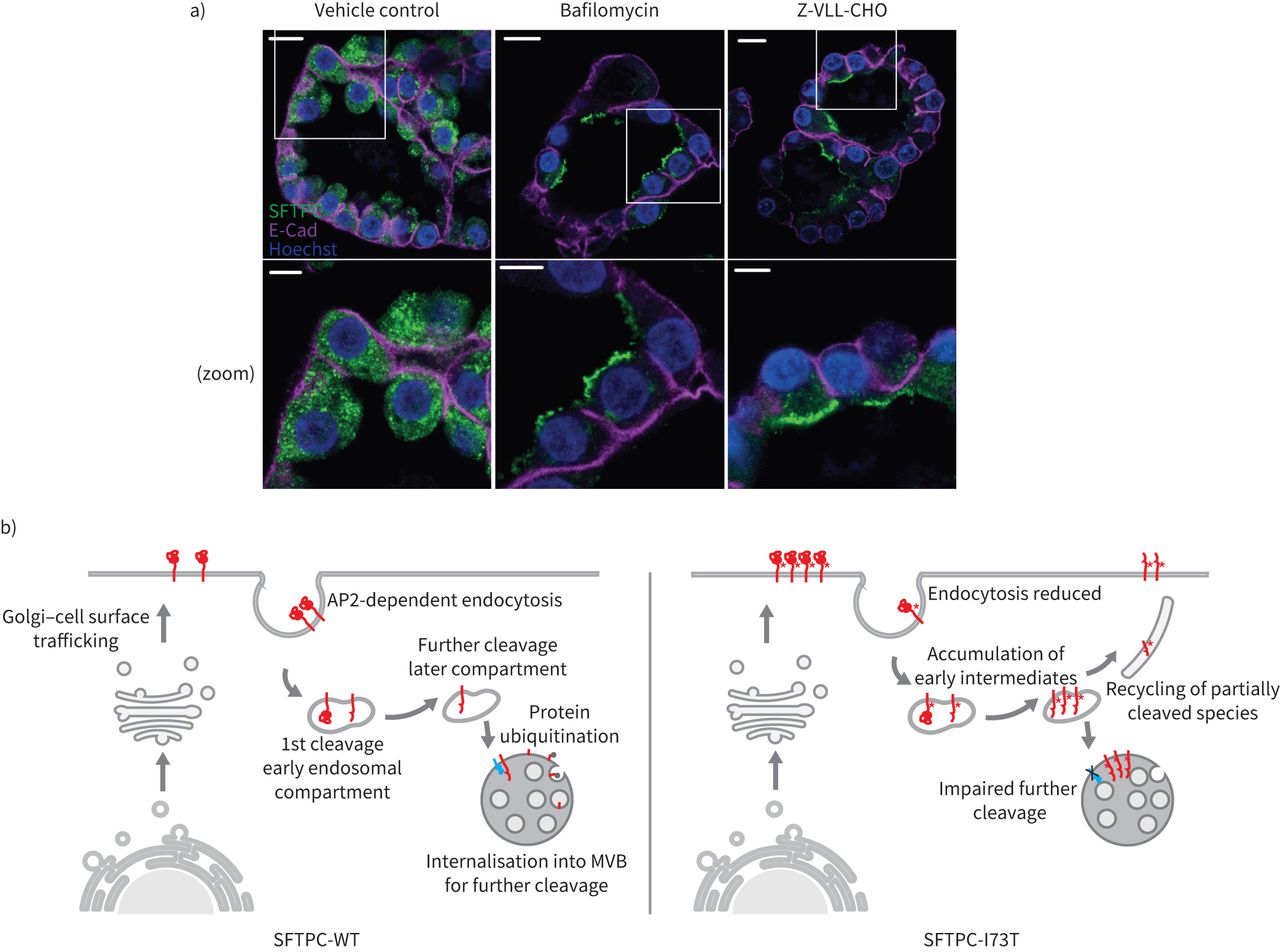
自从SFTPC表达只在at₂细胞,它是必要的,以验证这些发现在生理相关模型。因此我们生成瀑样从初级at₂隔绝人类肺细胞。这些肺瀑样由两极分化at₂细胞顶膜指向腔。免疫荧光显示内生SFTPC-WT点状的结构在顶端表面之下,虽然治疗bafilomycin(抑制CME)或Z-VLL-CHO(抑制endosomal sheddases)重新分配内生proSFTPC at₂顶端膜(图8)。这些发现与走私路线一致的机械化探索海拉细胞。
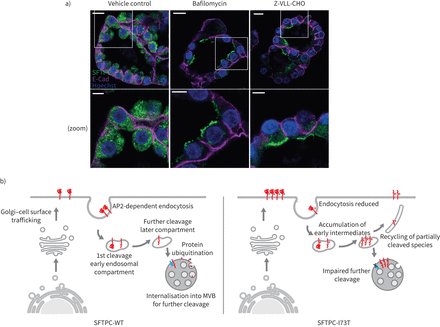
表面活性剂蛋白C (SFTPC)跟踪通过在人类肺泡瀑样细胞表面。人类肺泡)2型(at₂)细胞瀑样对待bafilomycin或Z-VLL-CHO SFTPC由免疫组织化学的本地化。钙粘蛋白(E-Cad)被用来描绘基底外侧膜。请注意,这两个化合物分配SFTPC顶端质膜。规模酒吧= 10μm或5μm(放大图片)。b)提出SFTPC跟踪模型。SFTPC pro-protein跟踪到质膜从高尔基体之前AP2-dependent clathrin-coated囊泡内吞作用。最初的c端裂发生在早期的内吞作用的舱前进一步乳沟膜蛋白酶在后面的车厢。这促进了泛素化,并允许掩饰成多泡体(多功能车辆总线)向前劈理和包装成片状的身体。I73T突变的存在,内吞作用降低,早期中间体积累和回收是由于后来块跟踪由失败后解理,泛素化和多功能车辆总线掩饰。
综上所述,这些实验表明,SFTPC经历蛋白水解作用远端连接域在核内体juxta-membrane乳沟,可能由一个膜结合sheddase。受损juxta-membrane乳沟,诱导药物或I73T突变,导致非生产性回收SFTPC核内体与质膜(图8 b)。
讨论
我们出发去理解机制SFTPC-I73T mistraffics在基维辛迪提供洞察其作用。在这一过程中,我们定义了野生型SFTPC的行程,其中包括贩运通过等离子体膜。我们观察到残留I73 SFTPC,位于细胞外链接器proprotein的地区,是贩卖作为其关键突变导致发展为多功能车辆总线和非生产性SFTPC受损细胞表面和早期核内体之间的循环。
异常保留SFTPC链接器区域与故障相关的泛素化,这并不影响内吞作用,但影响后续掩饰的SFTPC多功能车辆总线。我们的工作描述机制,解释了之前的观察,SFTPC-I73T本土化早期核内体,而野生型蛋白存在于多功能车辆总线(6]。链接器乳沟的确切机制导致多功能车辆总线针对野生型SFTPC尚不清楚,但链接器蛋白水解作用可能需要促进核内体和/或使K63-ubiquitination排序到晚。局部块贩卖SFTPC-I73T看到在培养细胞和SFTPC-I73T转基因小鼠(18]表明I73T产生一个不完整的块贩运和成熟的蛋白质。
SFTPC高度疏水性蛋白被认为促进肺表面活性物质的扩散与磷脂通过交互层(17]。基维辛迪SFTPC突变原因尚不清楚。老鼠缺乏SFTPC是可行的和正常生长39),而条件表达式的致病性SFTPC突变导致肺纤维化(18,40]。这表明发病机理是一种有毒的功能由异常蛋白质,而不是由于缺乏SFTPC损失函数。虽然BRICHOS域的突变导致ER应激(41,42),链接器区域突变体的毒性机理尚不清楚。
在这里,我们表明,SFTPC-I73T积累在质膜通过异常回收从核内体和受损的掩饰。回收SFTPC-I73T包括部分裂解中间SFTPC *,符合先前的质膜分析数据(19]。有趣的是,这种不正常的c端SFTPC碎片存在于支气管肺泡灌洗液的个人SFTPC-I73T突变(6,43]。集体观测,连同我们的发现表明,c端乳沟产品释放到细胞外空间在SFTPC异常的回收。人们很容易推测SFTPC-WT贩卖通过在正常细胞表面可能有作用at₂I73T摄动的突变细胞的功能。正在进行的工作将确定表面积累SFTPC多种形式,保留在回收核内体和/或c端乳沟发布产品本身影响at₂细胞内信号或与邻国。理解SFTPC及其处理的细胞内interactome中间体可能解释这新鲜。另外,饱和SFTPC可能有害的蛋白质回收途径对其他不相关的蛋白质的表面表达的影响。
在这个工作中,通过研究的天然致病变种SFTPC基维辛迪,我们发现所有野生型蛋白交通量通过多功能车辆总线的细胞表面,而不是直接从高尔基体。积累SFTPC-I73T变体的质膜因此沿着正常的路线,而不是mistrafficking反映迟钝本身。SFTPC拥有额外的功能是否需要运输通过细胞表面仍有待建立,可能理解的关键的角色SFTPC-I73T肺泡功能障碍导致基维辛迪。这样的洞察力也可能有助于澄清肺泡功能障碍在其他的角色,分家的IPF形式。
补充材料
补充材料
请注意:补充材料并不是由编辑部,编辑和上传已由作者提供。
补充材料和方法erj - 00267 - 2021. -补充
补充图S1erj - 00267 - 2021. - figure_s1
补充图S2erj - 00267 - 2021. - figure_s2
补充图S3erj - 00267 - 2021. - figure_s3
补充图S4erj - 00267 - 2021. - figure_s4
补充图S5erj - 00267 - 2021. - figure_s5
补充图S6erj - 00267 - 2021. - figure_s6
可共享的PDF
确认
作者要感谢玛格丽特·罗宾逊,马修·希曼保罗•雷纳Gershlick大卫和詹姆斯·埃德加(剑桥大学、剑桥大学、英国),安妮·范德做(肺学部门,莱顿大学医学中心,莱顿,荷兰)和迈克尔·比尔斯(美国宾夕法尼亚州费城宾夕法尼亚大学)有用的建议和试剂。
脚注
这篇文章一篇社论评论:https://doi.org/10.1183/13993003.02147 - 2021
可以从本文的补充材料www.qdcxjkg.com
利益冲突:J.A.狄更斯没有披露。
利益冲突:“卢瑟福没有披露。
利益冲突:美国阿伯没有披露。
利益冲突:J.E.室没有披露。
利益冲突:他们埃利斯没有披露规则。
利益冲突:a . van Schadewijk没有披露。
利益冲突:另外Hiemstra没有披露。
没有披露利益冲突:中华民国Marciniak博士审查。
支持声明:这项工作是由英国医学研究理事会(格兰特:先生/ S005552/1),但慈善信托,剑桥大学医院和NIHR剑桥生物医学研究中心(BRC)。资金信息,本文已沉积的Crossref资助者注册表。
- 收到了2021年1月28日。
- 接受2021年5月16日。
- 版权©2022年作者。
这个版本分布在Creative Commons归因执照的条款4.0。











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}