





摘要
2019冠状病毒病(COVID-19)大流行引起了科学界的迅速反应,阐明了严重急性呼吸综合征-冠状病毒-2 (SARS-CoV-2)引起的肺损伤的发病机制,并开发了有效的治疗方法。临床数据表明,严重的COVID-19最常见的表现为病毒性肺炎诱发的急性呼吸窘迫综合征(ARDS),这是在a型流感病毒诱发的肺炎背景下对临床实体机制了解最多的一种。与流感类似,高龄已成为发展严重COVID-19的主要宿主风险因素。在这篇综述中,我们将当前对SARS-CoV-2复制周期和宿主对COVID-19临床表现的反应的理解联系起来,借鉴了A流感病毒诱导的ARDS发病机制的概念,并讨论了这些观点如何影响我们对COVID-19诱导的ARDS的理解。我们还考虑了COVID-19和流感之间的重要差异,主要是COVID-19的多种临床表现和相关的淋巴细胞减少,干扰素-γ在介导宿主对这些病毒的免疫反应中的对比作用,以及SARS-CoV-2对血管内皮细胞的趋势性,评论流感作为COVID-19模式的潜在局限性。最后,我们探索了可以解释高龄与严重COVID-19易感性之间关联的衰老特征。
摘要
审查病毒ARDS发病机制,如何通知Covid-19的不断发展模型,以及老化的标志如何解释严重Covid-19的年龄相关的发病率和死亡率https://bit.ly/39ca0c0.
主要的小说理念和假设
我们的综述探讨了A流感病毒诱导的急性呼吸窘迫综合征(ARDS)作为理解2019冠状病毒病(COVID-19)诱导的ARDS发病机制和年龄作为严重疾病的危险因素的范式。我们整合了A型流感病毒诱导的ARDS病理生理学的现有知识,讨论了共享的临床结果如何将流感作为COVID-19的一个近似模型。我们认为,严重COVID-19受损的干扰素- i和-III反应与严重急性呼吸综合征冠状病毒(SARS-CoV)和流感病理生物学类似,表明这些病毒之间存在保守的毒力机制。我们还根据最新的临床数据回顾了这些病毒引起的免疫反应的临床明显差异,并建议研究人员在阐明SARS-CoV-2的致病性的持续努力中注意这些差异。具体来说,我们认为,严重COVID-19的高凝和高炎症状态是SARS-CoV-2向性扩大的结果,这使其能够感染血管内皮细胞,而细胞因子风暴生理学在较小程度上起到了作用。此外,我们还讨论了干扰素-γ活性在流感感染中的有害影响,以及这一观察结果如何与重症COVID-19的发现形成对比。我们还探索了与严重COVID-19相关的淋巴细胞减少的潜在病因:病毒的嗜性扩大,血清细胞因子(特别是白细胞介素-6和肿瘤坏死因子-α)升高,以及淋巴细胞过度聚集到肺部。最后,我们讨论了衰老的某些特征(表观遗传改变、线粒体功能障碍、端粒磨损、细胞衰老和细胞间通讯改变)如何使老年人更易患严重的COVID-19。我们推测,SARS-CoV-2影响t淋巴细胞和髓细胞生理的能力,加上年龄相关的适应不良的生物学现象,解释了老年与covid -19相关发病率和死亡率风险增加之间的强相关性。
介绍
一种新型冠状病毒(严重急性呼吸综合征-冠状病毒-2,或SARS-CoV-2)的传播挑战了世界各国政府的卫生保健系统和公共卫生政策的能力[1].截至2020年7月,2019冠状病毒病(COVID-19)大流行已导致超过1200万例病例和55万多例确诊死亡,尽管实施社会保持距离措施和戴口罩已减少了病毒在世界许多地区的传播[2].然而,它的高传染性预示着进一步的感染浪潮,需要继续努力充分阐明其发病机制和确定假定的治疗靶点。SARS-CoV-2感染导致的死亡率主要通过病毒性肺炎诱发急性呼吸窘迫综合征(ARDS)的发展而发生,ARDS是一种临床实体,在甲型流感病毒(流感)感染的背景下最容易理解[2那3.].虽然SARS-COV-2的确切机制导致ARDS以及某些宿主因子如何赋予发育严重疾病的风险增加仍然不明确,但有一个因素被出现为疾病严重程度和死亡风险的主要预测因素,这是年龄的4.那5.].事实上,意大利和中国在大流行早期的报告指出,年龄>80岁的患者的病死率为15-20%,而年龄小于50岁的患者的病死率小于1%。在这篇综述中,我们研究了流感诱导的ARDS病理生理学的临床相关机制,讨论了它们与COVID-19病理生理学的相关性,以及它们如何与COVID-19特异性临床结果进行对比,并探索可能导致年龄相关性严重COVID-19易感性的标志性生物衰老过程。
病毒复制循环和临床介绍
SARS-CoV-2(一种包膜的、阳性的单链RNA病毒)属于Betacoronavirus.属。它的RNA序列与引起2003-2004年SARS流行病的病原体SARS- cov有79%的相同[6.].在结构上,两种病毒在其包膜上含有穗状花序,其在其酶促活化之后via宿主蛋白酶和随后与人ACE2受体结合,介导病毒融合和内吞作用[7.].一旦进入细胞,病毒基因组就会被依赖病毒RNA的RNA聚合酶转录,然后被宿主核糖体翻译成合成病毒蛋白质。最后,成熟的病毒粒子聚集在细胞质中,在那里它们为胞吐作好准备[8.].
SARS-COV具有透明的气道上皮细胞和II型肺细胞的冠状动脉,其依赖于人ACE2受体,以及用于S蛋白裂解的宿主膜丝氨酸蛋白酶TMPRS2和随后的活化[7.].虽然SARS-COV-2分享了这种热衷和侵袭机制,但它还获得了葡萄牙裂解遗址,允许通过普遍存在的Furin蛋白酶进行S-蛋白质融合域暴露[9.].这些furin蛋白酶存在于全身无数同样表达ACE2的细胞的细胞膜上,包括血管内皮细胞[10.].这种扩大的趋向性最终转化为传染性的增加以及相对于SARS-CoV感染的新的临床结果。
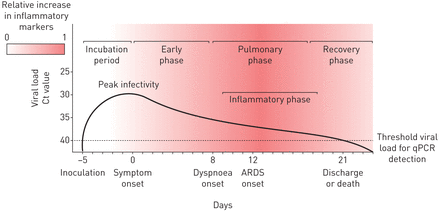
目前SARS-CoV-2的传播模型建立的平均潜伏期为5天,病毒载量在症状出现前达到峰值,之后单调下降。根据最新的报告,在鼻后/口咽拭子上至少8天和症状出现后平均20天内可检测到病毒载量[11.-14.].虽然病毒脱落的最大持续时间尚不清楚,但在大多数病例中,它可能在感染后40天减弱。在感染的早期症状阶段报告的症状差异很大,从轻微的身体症状,如发烧、疲劳、干咳和腹泻,到喉咙痛和味觉丧失;有些病例在本阶段仍完全无症状[15.-17.].在许多患者中,这些前驱症状随后是感染的肺阶段,其特征在于呼吸道症状和调查结果,包括呼吸困难,放射线肺浸润和低氧血症[14.].与COVID-19相关的大多数发病率和死亡率发生在以下炎症期,其特征是免疫反应失调和高凝状态,与危及生命的并发症有关,包括心、肾衰、脑血管疾病和ARDS [18.那19.].最后,在幸存者中,遵循恢复阶段,其中修复了炎症和受损的肺组织,最终恢复器官系统稳态(图1).在与炎症期相关的危及生命的后遗症中,ARDS是最常见的,占需要重症监护病房(ICU)住院的COVID-19患者的很大比例[15.].
目前对病毒ARDS和宿主免疫反应的理解
柏林标准通过急性事件(如呼吸道病毒感染)急性缺氧呼吸衰竭术语(如呼吸道病毒感染)在没有纯的肺部成像的肺部成像的情况下,通过急性发生的急性发生(如呼吸道病毒感染)定义ARDS。20.].ARDS是一种异质性的临床综合征,包括一系列需要进一步表征的内型,以发展有效的治疗方法[21.].在了解病毒性肺炎引起的急性呼吸窘迫综合征的病理生理学方面,特别是在流感的背景下,已经取得了很大进展[3.].对流感诱导的ARDS的规范理解从呼吸上皮细胞的细胞质中存在负面意义,单链RNA基因组(图2).细胞质病毒RNA触发I型和-III干扰素(IFN-1和-III)的分泌,通过激活诸如含量的受体,线粒体相关的抗病毒信号蛋白,以及NOD-,LRR-和含吡啶结构域的蛋白3炎炎症[22.那23.].这些抗病毒途径的诱导导致干扰素刺激的基因和核因子-κB-调节基因的上调,以及疗效和调节免疫细胞的募集。肺泡巨噬细胞通过吞噬感染和凋亡的上皮细胞,促进病毒间隙,分泌更炎症和趋化性细胞因子,包括IL-6和IL-8,促进巨噬细胞反应的特征[24.].这些肺上皮细胞和肺泡巨噬细胞反应协同募集募集其他免疫细胞类型,诱导中性粒细胞和天然杀伤(NK)细胞的侵袭以及将单核细胞的循环单核细胞分化为单核细胞衍生的巨噬细胞和树突细胞。

流感引起的急性呼吸窘迫综合征发病机制的关键组成部分流感的复制周期需要宿主上皮细胞的细胞质中存在病毒RNA,抗病毒途径如toll样受体、线粒体相关的抗病毒信号和NLRP3炎症小体能够识别病毒RNA。宿主细胞通过上调核因子(NF)-κ b调控基因(NF-κBrg)和分泌i型和iii型干扰素(IFN-I和IFN-III)作出反应,从而刺激干扰素刺激基因(ISGs)的转录。感染的上皮细胞来源的IFN-I和IFN-III诱导上皮细胞凋亡,白细胞介素(IL)-1β和IL-18分泌激活肺泡巨噬细胞,吞噬凋亡细胞。肺泡巨噬细胞也分泌IL-6和IL-8,将包括单核细胞在内的中央免疫系统召集到肺实质。单核细胞分化为单核细胞来源的巨噬细胞和树突状细胞。单核细胞来源的巨噬细胞也有助于被感染的上皮细胞吞噬,并分泌肿瘤坏死因子(TNF)-α和一氧化氮(NO)来激活上皮细胞凋亡。树突状细胞通过采集肺泡抗原并迁移到淋巴结,在那里它们将病毒抗原提供给naïve t细胞,从而连接先天和适应性免疫反应。经过抗原特异性克隆扩增后,t淋巴细胞被调动到肺中。CD8+当通过CD4共刺激时,T细胞在病毒抗原呈递细胞上施加细胞毒性+T细胞,其分泌II型干扰素(IFN-II),CD8的负调节剂+T细胞活性。反过来,CD4+T细胞通过调节T细胞(Treg)负调节,其也促进上皮修复via通过抑制肺泡巨噬细胞来源的IL-6和IL-8的分泌,增强其吞噬细胞活性(efferocytosis)来减轻炎症。NK:自然杀手。
作为先天免疫反应的一部分,中性粒细胞和NK细胞被招募到肺实质,以响应趋化细胞因子的分泌,包括CCL2、CCL5、CXCL8和CXCL10 [25.那26.].中性粒细胞诱导非特异性上皮细胞坏死via许多效应化合物的分泌,包括中性粒细胞胞外陷阱。虽然这些过程对流感病毒和其他病原体发挥保护作用,但如果不加以抑制,它们也会导致肺损伤[27.].类似地,在流感感染期间积极招募NK细胞,并施加Pro-坏死和-apofotic效应via细胞毒性和粒酶的穿孔素的分泌;的确,过度NK细胞介导的细胞毒性与从不受控制的肺损伤致死流感感染相关[28.].渗透单核细胞衍生的巨噬细胞和树突细胞通过分泌包括TNF-α和一氧化氮的其他促炎分子,通过诱导上皮细胞凋亡来驱动流感清除和肺泡损伤来有助于免疫应答29.那30.].树突状细胞还通过从肺泡中提取病毒抗原并迁移到引流淋巴结,在先天免疫系统和适应性免疫系统之间架起桥梁。在那里,它们作为naïve CD8的抗原提呈细胞+和CD4+T细胞,以抗原特异性方式激活它们以扩张和成熟,之后将它们动员到肺部[31.].曾经在肺部,CD8+T细胞诱导呈递病毒抗原的细胞的裂解,和CD4+t细胞以多种方式调节炎症反应,包括分泌额外的细胞因子,如IFN-II [32.那33.].
这些免疫过程导致流感病毒的主动间隙,以牺牲上皮细胞破坏和长期炎症的严重肺损伤为代价,表现为患者的副中的ARDS。有趣的是,事后剖析屈服于流感诱导ARDS的患者的研究表明,大多数患者中显示出不可检测的病毒载量,表明病毒的直接细胞毒性效应不是死亡率的唯一驱动因素;宿主无法抑制炎症和修复受损的肺组织有助于伟大程度[34.].这些发现对生理过程产生了大量兴趣,该过程介绍了肺损伤的炎症和修复的分辨率以及它们如何为病毒性肺炎诱导的ARDS相关的发病率和死亡率产生贡献。已经确定了许多负责调解恢复阶段的主机组件,包括Foxp3+调节性T细胞(Treg)[35.-42.].Treg细胞以其通过CD4负调控促进免疫自身耐受的作用而闻名+T细胞,但它们还可用于其他稳态功能,通过抑制巨噬细胞的促炎症作用和增强吞噬活动来介导炎症的分辨率[39.那42.那43.].Treg细胞还促进肺上皮细胞增殖viaAmphiregulin的分泌,表皮生长因子受体的一种配体,以及角质形成细胞生长因子[44.那45.].这些过程调节宿主免疫反应,并帮助恢复肺实质的稳态。因此,它们潜在的功能障碍部分解释了流感病毒引起的急性呼吸窘迫综合征患者的广泛结局。
洞察Covid-19诱发的ARDS
与Covid-19的临床介绍相关的异质性促使呼吸道疾病的新型范式的概念化,以解释Covid-19的观察到的可变性和个性化临床管理[46.].尽管如此,最近的队列研究报告称,多达85%的ICU与Covid-19患者符合ARDS的柏林标准定义,并且为低潮数和通风等领域的完善的支持性干预措施导致了显着的改善氧合和肺顺应性[47.].因此,在等待更多针对疾病的数据的同时,我们有理由探索病毒性肺炎导致ARDS的其他原因,以收集对严重COVID-19的见解。因此,有证据表明,流感和SARS-CoV-2感染之间既有相似之处,也有对比之处[48.].
SARS-CoV-2全球免疫特征
表征Covid-19的宿主免疫应答的早期作品表明,由血清细胞因子(特别是IL-1β,IL-6和肿瘤坏死因子(TNF)-α),受影响的干扰素反应和外周淋巴病症组成的免疫签名提出了一种免疫签名。严重疾病的标记;其他相关的炎症性血清标记物包括升高的铁蛋白水平,乳酸脱氢酶,D-二聚体,C-反应蛋白和凝血因子[4.那14.那49.].此外,SARS-COV-2和流感感染后肺上皮细胞的转录分析体外揭示了IFN-I和-III信号在宿主响应中的类似抑制这些病毒和共享细胞因子签名(包括IL-6和TNF-α)。相比之下,SARS-COV-2特异性免疫签名鉴定体外其特征是与流感相关的高IFN-II信号和趋化因子表达[50.].
IFN-I和-III响应受损
Covid-19和流感与相对于其他病毒的IFN-1受损的IFN-1和-III宿主反应有关,Covid-19严重程度与损伤程度相关[50.那51.].流感和SARS-CoV的基因组的非结构蛋白1(NS1),其拮抗IFN-I和-III信令的代码;类似地,SARS-CoV的-2蛋白ORF6,ORF8及其核衣壳还抑制IFN-I信号体外[52.-54.].NS1的保守性,加上相关的COVID-19 IFN-I和-III损伤,表明SARS-CoV-2与SARS-CoV具有相同的毒力因子,这些因子也在流感感染中重现。IFN-I调节巨噬细胞的促炎反应,使我们有理由假设IFN-I的下调可以解释与COVID-19相关的高炎症状态[55.].然而,在sars - cov -2感染的细胞中,药物抑制IFN-I信号体外没有增加细胞因子的产生,这表明受损的IFN-I分泌是不足以解释COVID-19 [的严重高炎症状态50.].IFN-III(或IFN-λ)与IFN-1共享类似的功能,但在不诱导过度炎症的情况下促进流感清除更有效[56.].IFN-III对于介导对流感的初始反应,在调节中性粒细胞的组织破坏性以及增强CD8中,持有非冗余作用+通过诱导树突状细胞迁移到引流淋巴结,记忆t细胞免疫对抗流感[57.那58.].这些观察结果使药术IFN-λ成为Covid-19的管理的候选者,尽管对IFN-λ对宿主对二次细菌感染的易感性的有害影响的鼠研究提出了对其安全的担忧[59.].然而,鉴于观察到这些促炎症分子能够限制SARS-COV-2的复制体外,正在进行的临床试验中确定的外源IFN-I和IFN-III给药治疗益处为COVID-19(ClinicalTrials.gov/NCT04343768和nct04354259.,分别)[60.].
Cytokine Storm作为Covid-19严重程度的司机
在应对SARS-CoV-2和流感时分泌的细胞因子的重叠可以解释为在两种病毒复制周期中宿主细胞的细胞质中存在病毒RNA,这可能诱导类似的细胞内抗病毒途径的激活,并随后将类似的免疫细胞招募到呼吸上皮。SARS-CoV-2的促炎免疫特征被比作巨噬细胞激活综合征(MAS),这是一种在自身免疫性疾病中观察到的危及生命的临床实体,并在许多病毒感染中被模仿,包括流感[61.那62.].MAS与NK细胞的细胞分解活性受损和特异性CD8有关+T细胞亚ppopopation,其任务是裂解感染的宿主细胞,以防止受损细胞长期分泌炎性细胞因子[63.].在细胞介导的裂解等损伤是由于高水平的IL-6的驱动时,建立的细胞因子驱动的促炎性细胞因子分泌的恶性循环。因此,升高的IL-6水平和它们与严重的疾病相关联被认为是反映了缺乏适当的调节和分辨率的过度旺盛的炎症反应,模仿一个MAS样病理状态。
Covid-19的血管病变和细胞因子风暴假设的影响
当试图使用流感相关的细胞因子风暴来得出关于Covid-19的潜在毒力机制时,SARS-COV-2的扩张的热衷于造成大量挑战。重要的是,屈服于Covid-19的患者的可用数据表明SARS-COV-2感染内皮细胞引起炎症(内皮炎)[64.].这一观察到的内皮炎支持了SARS-CoV-2对血管内皮细胞具有趋向性的观点,血管内皮细胞表达ACE受体[10.].此外,病毒的毒性可能在介导严重COVID-19比流感可以发挥更大的作用,因为事后剖析复制型病毒的检测是在后者[不太频繁34.那65.].这些发现也可以解释相关的多系统器官衰竭和hypercoagulable状态严重COVID-19,因为当地肺endothelialitis会激活的凝血级联和旺盛的生产endothelium-derived促炎细胞因子而不需要调用一个MAS-like病理状态。此外,与显示细胞因子风暴迹象的其他非COVID-19 ARDS队列报告的血浆IL-6水平相比,COVID-19患者报告的血浆IL-6水平似乎平均显著降低(10- 40倍)[66.].这些观察结果较低的假设可信度升高的血清细胞因子正在推动严重Covid-19中观察到前所未有的发病率和死亡率,这表明它们是局部血管病变的后果。值得注意的是,在没有可检测的利益的情况下,最近停止了探索Covid-19的单克隆IL-6受体抗体的治疗益处的临床试验(ClinicalTrials.gov/NCT04315298).
干扰素-II的二元角色
流感和Covid-19之间有关于IFN-II(或IFN-γ)在驱动疾病严重程度方面的作用的重要差异。以前关于IFN-γ在宿主对流感响应中的作用的研究表明,通过促进CD8的存活,IFN-γ信号传导的IFN-γ信号传导的衰减可以通过促进CD8的存活来改善临床相关结果+T细胞[67.那68.].低IFN-γ在流感中的这种保护作用不同于CD4产生的IFN-γ水平之间观察到的逆相关性+T细胞和Covid-19的严重程度[4.].此外,SARS-CoV-2可诱导更强的IFN-γ反应体外这表明IFN-γ可能在宿主对COVID-19的免疫反应中发挥更大的作用[50.].一些人认为,il -6- ifn -γ比值可以作为一种临床工具,根据患者发展为严重COVID-19的相对风险对患者进行分层,对该比值作为严重COVID-19风险分层措施的初步分析显示,结果很有希望[69.].然而,在其部署之前,需要更好地了解IFN-γ在驱动严重疾病中的作用以及较大群组中的IL-6-To-IFN-γ比的验证。
Covid-19相关淋巴蛋白
严重的Covid-19与淋巴细胞减少有关,淋巴细胞减少影响T-而不是B细胞舱[4.].类似的淋巴细胞计数变化也发生在其他病毒感染中viaTNF-α介导的淋巴细胞再循环抑制的机制[70].事实上,IL-6抑制改善了COVID-19患者子集中严重疾病的淋巴细胞减少,表明血清细胞因子升高在耗尽外周血t淋巴细胞中发挥了作用[71.].尽管如此,Covid-19患者最近的免疫性分析表明,T细胞隔室虽然严重影响更严重与与非Covid-19肺炎病例相比,在Covid-19患者的外周血中仍然显着减少了Covid-19患者的显着减少[72.].该发现表明,细胞因子过量不是Covid-19淋巴细胞增长的唯一因果因素。对Covid-19相关淋巴细胞增长的另一种潜在的解释是肺中淋巴细胞的过度招募和封存。支持这个假设,事后剖析检查Covid-19患者的独立队列鉴定在病例子集中鉴定肺淋巴浸润,并在患有轻微和严重的Covid-19患者的支气管肺泡灌洗液样品中进行类似的观察[64.那73.那74.].最后,一些hypothesise这种淋巴细胞是SARS冠状病毒-2,这可能赋予细胞毒性T细胞群的扩大取向的直接后果;然而,这样的取向仍然没有得到证实。由于淋巴细胞正在考虑患者的危险分层,阐明的病因COVID-19相关的淋巴细胞是我们COVID-19发病机理和努力,发展成严重的疾病[风险识别患者的理解至关重要75.].
老化驱动器严重Covid-19的潜在机制
高龄与发展危及生命的感染的风险增加有关[78.].事实上,几乎90%的流感相关死亡率和不成比例的covid -19相关死亡率发生在>65岁的人群中[4.那5.那79.那80].这种与年龄相关的严重疾病易感性并不仅仅是因为长期暴露于环境和宿主风险因素,如烟草烟雾或原发性高血压,也通过发展不良的生理过程,影响个体在面对应激源时维持体内平衡的能力。这些过程被认为是衰老的标志,并与年龄相关的压力易感性(也被称为同种狭窄)广泛相关。81.那82.].全球人口日益老龄化及其众多与年龄相关的共病,使人们对了解这些衰老特征的病理生理学产生了浓厚兴趣,特别是在它们与严重COVID-19风险相关的情况下。虽然它们的影响尚未在SARS-CoV-2感染的背景下阐明,但我们目前对它们在流感中的作用的了解,为了解为什么衰老是严重COVID-19的发展的一个风险因素提供了见解(图3).
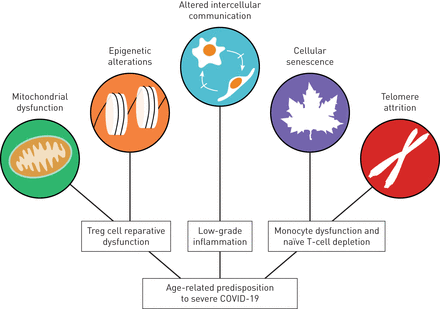
老龄化特征对严重2019冠状病毒病(COVID-19)的年龄相关易感性的贡献的例子。年龄相关性线粒体功能障碍可诱导调节性t细胞(regulatory t cells, Treg)表观遗传改变,损害其促恢复功能,阻碍炎症的正确解决和肺损伤的修复。单核细胞和naïve t淋巴细胞在持续复制的端粒磨损后会发生细胞衰老,削弱宿主对病毒挑战的有效免疫应答能力或对疫苗产生记忆性t细胞应答的能力。最后,细胞间通讯的改变是与衰老相关的低级别炎症的基础,这有助于年龄相关共病的发展。
表观遗传改变和线粒体功能障碍
晚期的年龄与免疫细胞的表观遗传和代谢景观的变化有关,其介导对病毒肺炎诱导的ARDS的宿主反应,包括Treg细胞[39.].Treg细胞的促修复功能与调节性T细胞谱系特异性细胞基因座的DNA低甲基化相关联。事实上,DNA甲基转移酶的抑制Treg细胞加速肺损伤分辨率在鼠流感模型,表明DNA甲基化是必需的,以保持对肺损伤后的寿命,但也妨碍Treg细胞修复功能的Treg细胞谱系[40.].当老化时显示出Treg细胞功能和数量的改变时,发生这些变化的机制仍然不清楚;然而,最近的研究表明它们可以通过与老化的线粒体功能障碍引起的有毒代谢物和活性氧物种的积累来介导的[83.].例如,代谢物在细胞内的积累L -2-羟基凝集在其缺氧期间的合成和线粒体功能障碍的其他状态诱导DNA高甲基化并损害Treg细胞中的抑制功能[84.].这种代谢物的积累最终可以通过抑制Treg细胞前分解和前修复途径相关关键位点的表达来抑制Treg细胞依赖性的ARDS恢复。考虑到线粒体功能障碍与年龄相关的现象,无论其近因如何,类似的现象也可能发生在病毒性ARDS解决所需的其他细胞类型上。
端粒耗损和细胞衰老
衰老与细胞连续复制引起的染色体端粒侵蚀(端粒磨损)和体细胞(包括单核细胞和淋巴细胞)伴随的细胞周期阻滞(细胞衰老)有关。老年小鼠的单核细胞无法正常吞噬细菌病原体,但相对于年轻小鼠,其产生的TNF-α水平显著升高。这些发现与单核细胞的端粒长度相关,单核细胞的端粒长度在老年人中较短,提示在病原体清除过程中存在与年龄相关的损伤,以及部分由端粒磨损介导的TNF-α介导的过度凋亡[85.].这些与年龄相关的现象也会影响幼稚T-和B细胞数,表现为老宿主中这些细胞的低外周血计数[86.].随着胸腺退化的进程,与年龄相关的naïve T细胞计数的减少对适应性免疫系统产生抗原特异性记忆淋巴细胞的能力构成了严重的障碍[87.].因此,遗传因素最近全基因组关联分析链接到识别的风险等位基因与CXCR6表达,这在组织驻留记忆T细胞[表达的趋化因子受体的水平降低相关的严重COVID-19敏感性88.].这一发现支持以下假设:受损的记忆T细胞功能易患发展为严重COVID-19。年龄相关的幼稚T细胞缺陷也假说认为起到住院时间延长一个角色,在受流行性感冒引起的急性呼吸窘迫综合征的人口老龄化易感性增加严重疾病[89.].最后,NaïveT细胞耗尽也使季节性流感疫苗的疗效降低,随着年龄的增加,限制了老年成人的预防治疗方法[80].
改变的细胞间通信
与衰老相关的复制性衰老影响多种体细胞,并与促炎细胞因子的基础、慢性分泌有关,这种现象被称为衰老相关分泌表型(SASP) [82.].根据衰老假说,在整个衰老过程中,这些sasp样细胞的积累导致效应免疫细胞的持续招募和激活,扰乱免疫系统的促炎臂和抗炎臂的局部通讯,诱导组织损伤,阻碍组织修复[90.].因此,选择性耗竭应激诱导的衰老细胞在体内降低急性肺损伤后sasp相关转录信号,改善小鼠的生理功能,提示衰老肺泡上皮细胞的积累最终损害了宿主从急性肺损伤中适当恢复的能力[91.].这种促炎状态也可能解释许多年龄相关疾病的易感性,包括原发性高血压,这是与个人发展严重COVID-19风险相关性最强的共病[92.].
结论
我们对COVID-19的不断理解表明,流感发病机制为了解COVID-19提供了思路,特别是关于受损的IFN-I和-III宿主反应。然而,流感的发现不足以解释重症COVID-19的独特临床过程、内皮炎、IFN-II的作用以及与重症COVID-19相关的淋巴减少。我们认为,这两种感染之间的这种差异代表了sars - cov -2特异性的毒力机制,如果得到阐明,可能会为新的治疗策略的发展提供信息。因此,我们认为,这些差异需要加以解释,并应成为正在进行的研究工作的重点。我们还推测,严重的COVID-19特异性免疫特征,加上与年龄相关的适应不良现象,可能会阻碍宿主抑制严重COVID-19炎症期和解决肺损伤的能力,导致了在老年人中延长ICU护理事件的发生,以及伴随而来的与延长ICU时间相关的发病率和死亡率。考虑到这些老龄化特征的相互依存和协同性质,我们建议,应该共同研究这些特征对严重COVID-19的相对易感性的影响。最终,衰老生物学和严重病毒性肺炎诱发的ARDS相对易感性之间的机制关联具有实质性的临床意义,在未来制定针对人群的COVID-19预防和治疗干预措施时需要仔细考虑。
可分享的PDF.
脚注
兴趣冲突:M.A.托雷斯Acosta没有披露。
利益冲突:B.D. Singer有一项美国专利App. 15/542,380,“加速解决急性肺炎症的成分和方法”正在申请中。
支持声明:M.A. Torres Acosta获得NIH T32GM008152奖支持。B.D. Singer获得NIH奖K08HL128867、U19AI135964、R01HL149883和P01AG049665的支持。本文的资金信息已存入Crossref资助者注册表。
- 收到2020年5月28日。
- 接受2020年7月23日。
- 版权所有©ers 2020
此版本在Creative Commons归因非商业许可证4.0的条款下分发。











![Stereotypical clinical course of coronavirus disease 2019 complicated by acute respiratory distress syndrome (ARDS). Mean incubation period and viral load threshold cycle (Ct) values at days 0, 7, 14 and 21 according to He et al. [12]. Median days from symptom onset to dyspnoea (interquartile range 4–9 days), ARDS (interquartile range 8–15 days), and discharge or death (interquartile range 17–25 days) according to Zhou et al. [14].](http://www.qdcxjkg.com/content/erj/56/3/2002049/F1.large.jpg?width=800&height=600&carousel=1){kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}