





摘要
背景在法国,Mepolizumab作为严重嗜酸性粒细胞性哮喘患者早期获取计划的一部分可获得临时使用许可[临时使用授权](nATU))。本研究旨在分析在nATU中接受mepolizumab治疗的患者。
方法这项回顾性观察性研究分析了治疗开始后24个月患者的医院医疗记录数据。研究目标是描述随访期间患者的基线特征、疾病严重程度的演变和治疗调整;安全性也进行了调查。
发现总的来说,146例接受≥1剂量mepolizumab的患者被纳入研究。纳入时,患者平均年龄为58.2岁,严重哮喘平均持续时间为13.4年,37.0%患有呼吸道过敏。基线时,每位患者平均每年经历5.8次恶化,其中0.6次和0.5次分别需要住院治疗和急诊科就诊。在随访24个月时,这些值分别改善至每名患者每年0.6、0.1和0.1次恶化。大多数患者(92.8%)在基线时使用口服皮质类固醇,而24个月随访时为34.7%。此外,平均血液嗜酸性粒细胞计数从722个细胞·µL改善−1基线浓度为92细胞·µL−1随访24个月;肺功能和哮喘控制也有相似的趋势。
解释结果证实了临床试验的发现,证明mepolizumab与几个有临床意义的结果的重要改善相关,并且在临床试验控制环境之外的严重嗜酸性粒细胞性哮喘人群中具有良好的安全性。
摘要
Mepolizumab与几个有临床意义的结果的改善相关,并在临床试验控制环境之外的严重嗜酸性粒细胞性哮喘人群中显示出良好的安全性https://bit.ly/3bckeQ3
介绍
哮喘是一种常见的呼吸系统疾病,影响全球约3.6亿人,估计占法国人口的3.5-10.3% [1,2]。一小部分哮喘患者患有严重哮喘[3.],由几种临床不同的表型和内型组成[4- - - - - -7]。严重嗜酸性粒细胞表型的特征是持续嗜酸性粒细胞炎症,肺功能和哮喘控制下降,尽管使用高剂量吸入糖皮质激素(ICS)、其他控制剂和慢性或反复使用全身糖皮质激素,但复发性加重[4,8]。
Mepolizumab是一种抗白介素-5单克隆抗体,可选择性抑制嗜酸性粒细胞炎症[9]并被批准作为严重嗜酸性粒细胞性哮喘患者的附加治疗方法[10- - - - - -12]。随机对照试验(rct)表明,与安慰剂相比,mepolizumab可降低急性发作率,降低口服皮质类固醇(OCS)依赖性,并改善肺功能、哮喘控制和健康相关的生活质量[13- - - - - -16]。虽然随机对照试验的数据可以对治疗的临床疗效和安全性提供重要的见解,但这些研究通常是为了满足一个特定的主要目标,如评估OCS剂量或加重率的变化。此外,由于合格标准狭窄,随机对照试验可包括有限的患者群体,这不能反映一般哮喘人群[17]。因此,在正式临床试验的限制之外获得治疗效果的数据也很重要。
Mepolizumab于2015年12月在欧盟被批准用于严重嗜酸性粒细胞性哮喘患者[10]。在2018年2月mepolizumab上市之前,法国的患者已经获得了mepolizumab,作为早期准入计划(提名)的一部分临时使用许可[临时使用授权](nATU)),后来由安全炸药Sociale[18]。nATU仅限于因疾病严重程度而被认为无法等待商业化的患者。双方之间建立了协议国家代理Sécurité du Médicament et des producits de Santé(ANSM)和制造商(葛兰素史克),后者规定了患者监测程序,收集与疗效和安全性以及实际使用条件有关的数据。为了了解典型的患者途径并描述在现实环境中接受早期mepolizumab治疗的患者的特征,分析了nATU期间收集的数据以及从患者病历中回顾性收集的数据。目的是描述nATU患者的特征,并描述治疗开始后24个月的疾病严重程度演变和治疗调整。
方法
研究设计与处理
这项回顾性观察性研究(GSK ID: 207943;HO-17-18317)纳入了在法国开始mepolizumab治疗(100 mg皮下注射4周)的严重嗜酸性粒细胞性哮喘患者的医院医疗记录数据,作为nATU的一部分。nATU方案规定的医疗数据收集时间为2015年6月9日至2016年3月2日(nATU期间),以及2016年3月2日至2018年2月(nATU后),这允许在治疗开始后长达24个月的回顾性随访期。在nATU期间和之后记录的数据也包括在内,但不是nATU协议强制要求的。这项研究于2017年12月21日向卫生研究、研究和评估专家委员会宣布,并于2018年1月8日向国家数据保护和自由委员会宣布符合参考方法MR003。
参与医疗中心
参加的医疗中心在nATU有3名以上的病人,并同意参加。本研究中涉及的大多数肺科都是在大学医院(进一步详情见补充第1条)。
病人
纳入nATU的患者在参与中心接受了≥1次mepolizumab注射,由医生填写的治疗访问表证明。连续两次注射mepolizumab之间的所有延迟均为4±1周。为了证明mepolizumab的申请并帮助告知ANSM和葛兰素史克的后续验证,医生被要求证明:患者患有严重的嗜酸性粒细胞性哮喘(没有嗜酸性粒细胞性肉芽肿伴多血管炎的特征);目前没有其他合适的治疗方案;纳入RCT是不可能的;患者的临床健康状况需要紧急改变治疗,以避免严重恶化和/或严重的类固醇副作用。治疗获取表格包括血液嗜酸性粒细胞计数、加重率、症状控制和OCS剂量的信息。没有严格的资格标准来允许请求得到葛兰素史克的验证,而且患者必须愿意透露他们的个人医疗记录。
终点和评估
主要目的是描述纳入nATU的患者的概况,使用nATU内收集的额外数据。在mepolizumab开始前的12个月内评估基线特征,包括:哮喘持续时间;吸烟史;地理本地化;并发症;就业状况;asthma-induced残疾;社会经济地位;补充健康保险状况;与哮喘相关的恶化次数,包括需要住院或急诊科(ED)就诊的病例; atopic status; blood immunoglobulin (Ig)E, eosinophil and neutrophil levels; OCS dose; previous treatment adherence (estimated by investigators); forced expiratory volume in 1 s (FEV1);和FEV1/用力肺活量(FVC)比值。哮喘加重被定义为疾病恶化,需要急诊就诊、住院和/或使用OCS≥48小时或OCS日剂量增加≥50%。通过≥1个阳性皮肤点刺试验或过敏原特异性IgE血液试验(IgE水平>0.1 UI)确定特应性状态。
次要目标是描述随访期间(mepolizumab起始后≤24个月)疾病控制和治疗改变的演变。为了评估这些,我们检查了:病情恶化的次数以及如何处理(如。是否需要OCS、急诊科就诊和/或住院);FEV1;FEV1/ FVC比率;mepolizumab停药日期和原因(如果适用)。通过纳入物(<300、300 - <500、500 - <700和≥700细胞·µL)血液嗜酸性粒细胞计数分析哮喘加重率和OCS使用/剂量−1),并分析评估治疗前12个月患者对mepolizumab的不同反应水平(基于恶化率降低≥50%和OCS剂量降低≥50%)(补充章节2和3)。安全性终点包括不良事件、严重不良事件(SAEs)和相关不良事件的发生率。
统计分析
平均哮喘加重率报告为每位患者每年发作;使用泊松回归模型分析加重率的演变。对FEV进行趋势分析1, fvc, fev1/FVC比值数据、哮喘控制试验(ACT)评分和血液嗜酸性粒细胞计数在纳入和随访期间采用重复测量的混合线性模型。使用Kaplan-Meier方法估计mepolizumab治疗的持续时间。使用SAS软件(版本9.4 SAS institute Inc., Cary, NC, USA)进行统计分析。
结果
患者人群
在纳入nATU的160例患者中,来自20个参与中心的146例(91.9%)接受≥1次mepolizumab注射纳入本研究;13例(8.1%)患者未接受mepolizumab治疗,被排除在外(补充图S1)。一名患者在等待肺移植期间接受mepolizumab治疗严重慢性阻塞性肺疾病,被科学委员会认为不合格,除安全性分析外,被排除在所有分析之外。总体而言,61例患者(41.8%)进行了103次注射,延迟4周。这些延迟的原因仅记录了19次注射,包括度假、亲属死亡、与mepolizumab无关的健康问题以及患者忘记了。
基线人口统计学和临床特征概述表1。在62例确诊过敏的患者中,54例(87.1%)对空气过敏原(花粉、皮屑、霉菌、蟑螂)过敏,12例(19.4%)对食物过敏,5例(8.1%)对皮肤过敏。几乎所有患者(93.8%)有≥1种共病;最常见的是耳鼻喉疾病(56.2%)、心血管疾病(35.0%)及胃食管反流病(38.7%)(表1)。此外,38.7%的患者患有鼻息肉,17.5%的患者患有过敏性鼻炎,16.1%的患者患有阿司匹林加重的呼吸道疾病。此外,大多数患者(92.8%)在纳入时接受了OCS(平均每日剂量20.6±16.5 mg泼尼松龙当量),65.9%之前接受过omalizumab (表1)。
在mepolizumab治疗开始前的12个月,平均每位患者每年发作5.8次,其中0.6次和0.5次分别需要住院治疗和急诊科就诊(表2)。基线时的平均嗜酸性粒细胞计数为722细胞·µL−1相当大比例(n= 115,86.5%)的患者报告哮喘对他们的日常活动有显著影响。
随访数据
患者在治疗开始后平均随访8.4次;由于部分患者很少返回医院,最后一次就诊日期超过24个月,平均随访时间为24.2个月。连续注射mepolizumab,平均间隔4.2周。随访期间,共有48例患者停用mepolizumab,大多数患者报告缺乏疗效或缺乏与不良事件相关的疗效(n=29) (补充表S1)。第一次注射3个月后,91%的患者仍在接受mepolizumab治疗;6个月后下降到81%,随后在12个月和24个月下降到69%和66%。
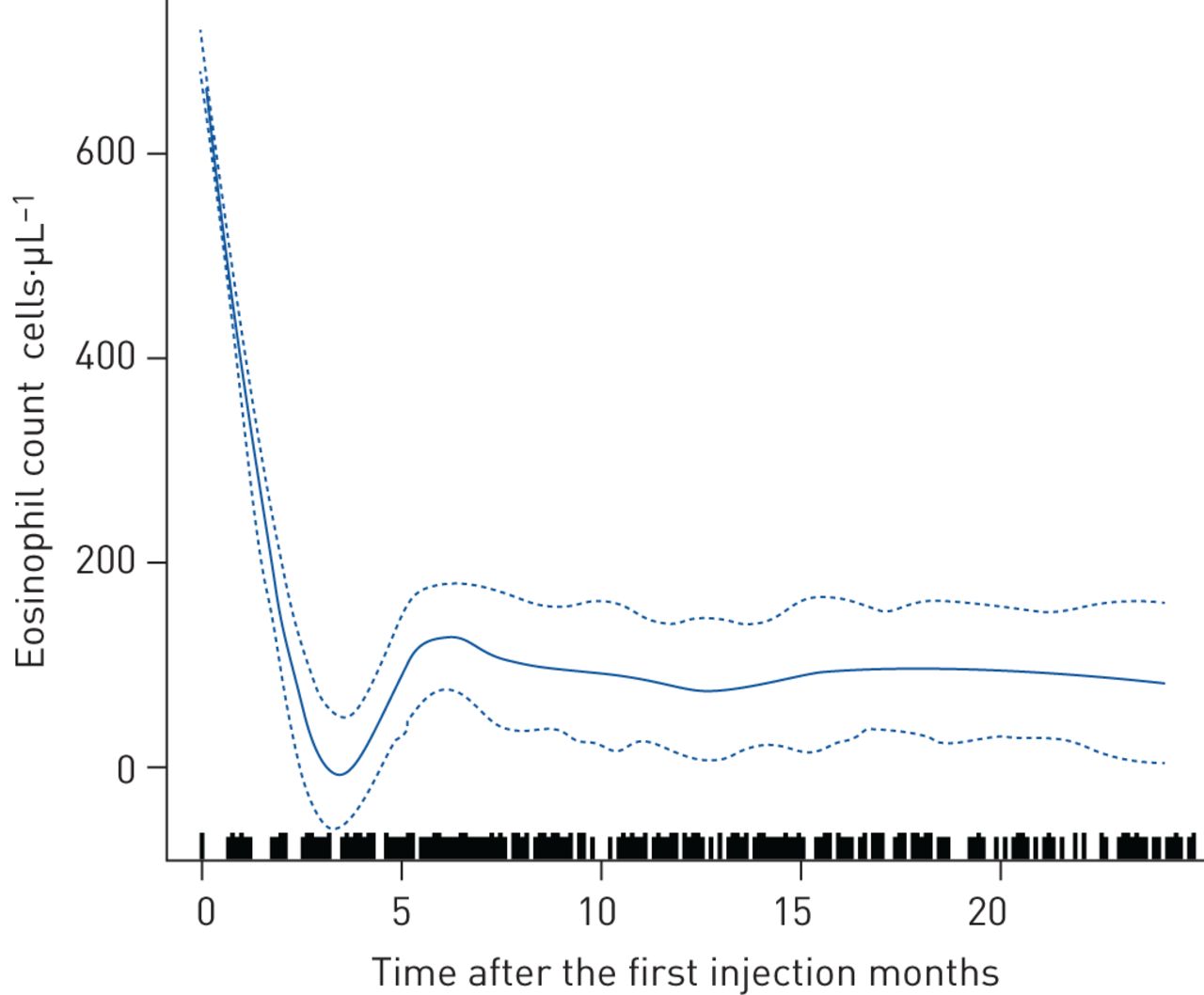
治疗期间,总急性加重以及需要住院和急诊科就诊的患者的平均急性加重率低于基线(表2);这一趋势与纳入时的血嗜酸性粒细胞计数无关(补充表S2)。与基线相比,随访期间使用维持性OCS的患者较少(基线时为92.8%)与12个月和24个月时分别为41.1%和34.7%),而仍在使用OCS的患者所需剂量较低(图1);当数据按照纳入时的血液嗜酸性粒细胞计数进行分层时,也看到了类似的趋势(补充图S3和补充表S3)。平均%预测支气管扩张剂前FEV1改进的与所有随访时间点的基线;意味着FEV1在治疗的前10个月,稳定上升至约70%的预测值,然后稳定(图2一个)。意味着FEV1/FVC比值在治疗的前10个月也有所增加,然后趋于稳定。mepolizumab治疗3个月后,平均ACT评分为17.4分;这超过了与基线(10.2分)相比≥3分的最小临床重要差异(MCID),并且在整个研究期间反应持续(图2 bc).均数±sd基线血嗜酸性粒细胞计数(细胞·µL−1)分别从3个月(722±500.0)、12个月(75±63.7)、24个月(92±72.3)下降至101±83.9 (图3)。
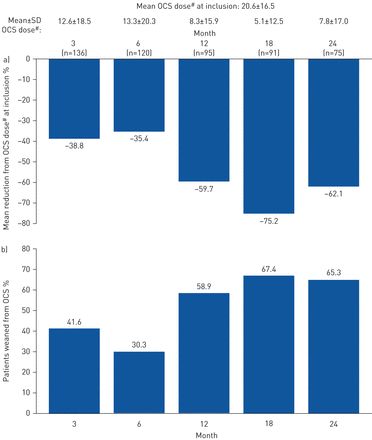
a)平均口服皮质类固醇(OCS)剂量变化;b)随访期间未接受维持性OCS的患者比例。#: mg强的松龙当量。基线时,8例患者数据缺失。随访12个月和24个月时,42名和31名患者停止治疗,8名和40名患者分别没有随访。
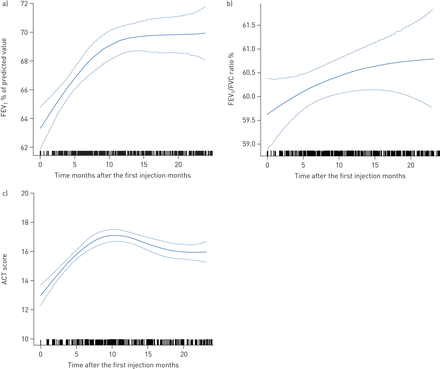
a) 1 s内预测支气管扩张剂前用力呼气量(FEV)百分比的变化1) b) FEV1/强迫肺活量(FVC)比值和c)哮喘控制试验(ACT)评分。连续的线代表每个分数的演变;虚线表示置信区间。
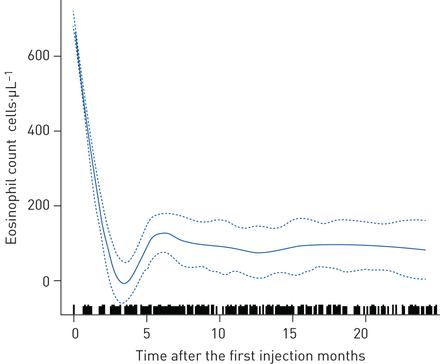
随访期间嗜酸性粒细胞计数的变化。连续的线代表每个分数的演变;虚线表示置信区间。
安全
在研究期间,100名患者报告了276起药物警戒事件。其中,103例与药物滥用有关(所有报告的给药间隔都不正确);根据患者的医疗记录,有173例被确定为可能与mepolizumab相关的不良事件。99例患者共报告159例非严重不良事件;41例患者因这些事件停用mepolizumab(29例报告“药物被认为无效”)。最常见的可能与药物相关的不良事件(n≥5%)包括:药物被认为无效(n=31);头痛(n = 14);衰弱(n = 12);哮喘(n=10) (表3)。相关不良事件包括5个系统器官类(SOC)事件“感染和侵害”,5个“血管紊乱”事件,2个与“过敏和非过敏反应”有关的事件以及1个“局部注射部位反应”事件(表4)。共有8例患者报告了14例可能与药物相关的sae;最常见的是哮喘(n=3例)。在本研究中,没有患者出现严重的全身反应、严重的心脏不良事件或肿瘤。报告了一例死亡(由哮喘加重引起),医生认为与mepolizumab无关。
讨论
提早入场计划(如。nATU)允许不符合严格的rct资格标准,但仍可能受益于mepolizumab治疗的患者在其商业化之前获得该药物。此外,从这些项目中收集的数据可以为在与现实生活非常相似的患者群体中更广泛地使用mepolizumab提供见解[19]。在这里,我们使用nATU登记的严重嗜酸性粒细胞性哮喘患者的数据调查了mepolizumab的有效性和安全性。我们发现美泊珠单抗与几种临床益处相关,包括临床意义上的急性发作和每日OCS剂量的降低,这与分别评估这些结果的两项随机对照试验的结果一致[14]。
我们在nATU患者中确定了一些严重疾病的指标,其中包括高的年度恶化率,大多数患者需要维持OCS,并经历了相当大的疾病负担。这些患者的疾病严重程度一般高于随机对照试验的患者。例如,参加MENSA和MUSCA III期研究的患者在筛查前平均每年发作2.7-3.8次(与5.8在本研究中)[13,15]。此外,本研究纳入时需要维持OCS的患者比例(92.8%)高于MENSA和MUSCA研究(23-27%)[13,15]。这些发现并不出乎意料,因为nATU中的患者在mepolizumab上市之前有合理的需求。与MUSCA试验相比,nATU人群中伴有鼻息肉的晚发型嗜酸性粒细胞性哮喘的比例似乎也过高(nATU患者中38.7%)与MUSCA为17-21%)[15]。尽管如此,这项研究中的疾病严重程度与现实世界中接受omalizumab患者的研究相似[20.和其他早期获取项目,包括法国的omalizumab和dupilumab ATUs [21,22]。
预防哮喘加重仍然是哮喘患者的重要治疗目标[23]。在本研究中,随访12个月和24个月后,我们观察到急性加重率比基线降低了86.2%。在MENSA (MEpolizumab作为重度哮喘患者的辅助治疗)和MUSCA (MEpolizumab辅助治疗重度嗜酸性粒细胞性哮喘患者)中,MEpolizumab (100 mg SC)使用32周和24周后,加重率分别降低了53%和58%。与安慰剂(14,15]。基于真实数据的其他研究也将mepolizumab与恶化的减少联系起来[24- - - - - -27];在最近的一个例子中(n=25), 82.6%的患者在治疗期间病情恶化较少,47.8%的患者在治疗期间病情没有恶化[24]。当我们分析纳入时的血液嗜酸性粒细胞计数数据时,所有亚组的加重率和OCS使用/剂量均有所降低(尽管样本量较小)。有趣的是,考虑到研究人群的疾病严重程度,需要住院治疗的病情加重率很低。这可能是由于大多数患者是在专门治疗严重哮喘的大学医院招募的,在那里患者通常有促进疾病管理的行动计划。
我们发现,大约33.0%和62.5%的接受mepolizumab治疗的患者在6个月和12个月时的每日OCS剂量减少≥50%(补充第2节)ontero -Perezet al。[24]也报道了大约60%的患者在治疗12个月后经历了OCS剂量的减少(尽管这并不限于减少≥50%)。值得注意的是,在SIRIUS (mepolizUmab类固醇减少研究)随机对照试验中,54%的患者在接受6个月的mepolizUmab治疗后,观察到每日OCS剂量比基线减少≥50% [14]。虽然这比我们的研究中患者的比例更大,但在现实环境中,OCS剂量的降低通常比rct中遵循的降低滴定方案更缓慢。因此,在我们的研究中,OCS的减少可能比SIRIUS进行得更慢,这由FEV的峰值改善所支持1在nATU中mepolizumab治疗6个月后的ACT评分。此外,在随访24个月时,我们观察到OCS使用/剂量的复发。然而,这可能是由于时间点之间患者数量的差异(18个月;N =91, 24个月;n = 75)。
在安全性方面,我们发现在24个月的研究期间,头痛和哮喘是最常见的不良事件,过敏、非过敏和注射部位反应的发生率较低。这些结果与随机对照试验的结果一致[14,15],并表明mepolizumab在更接近现实生活的环境中具有良好的耐受性。在nATU期间,48例患者停用mepolizumab,其中11例报告缺乏与不良事件相关的药物疗效。观察到的高停药率(32.9%)可能是由于mepolizumab在研究时是一种新产品,没有市场批准,因此没有可用的停药规则或指南。因此,如果几个月后没有看到任何好处,医生可以停止治疗。
本研究的主要局限性是数据收集和分析的回顾性性质。值得注意的是,大约40-60%的患者的ACT评分数据缺失,尽管我们在有数据的患者中确实观察到ACT评分较基线的变化超过了MCID。在本研究中停用mepolizumab的患者(由于不良事件或缺乏疗效)不包括在安全性和有效性结果中;由于这些措施取决于参与分析的患者数量和治疗持续时间,因此应谨慎解释所报告的数据。此外,本研究使用的医疗数据中没有记录ICS基线剂量;因此,这些不能包括在我们对nATU患者群体的基线特征和进化的评估中。结果也可能受到各种混杂因素的影响,例如,由于生物给药所需的定期与医疗保健专业人员接触,或以前接受过另一种生物治疗的患者,导致更严格的依从性(如。omalizumab)。最后,根据nATU的早期治疗标准,本研究中的患者患有特别严重的疾病,并在大学医院接受治疗,具有管理严重哮喘的专业知识。因此,我们的数据可能不能反映整个严重哮喘人群,特别是那些在这种环境之外接受治疗的人群。
尽管如此,这些来自在法国早期治疗项目中接受mepolizumab治疗的严重嗜酸性粒细胞性哮喘患者的数据证实了mepolizumab在降低急性加重率和OCS依赖性以及改善肺功能方面的有效性,这与现实生活中的使用非常相似。此外,安全性发现与临床试验中观察到的结果一致。虽然还需要更多的研究来全面评估mepolizumab在真实世界的长期临床实践中的安全性和有效性,但这项分析为正在考虑治疗严重嗜酸性粒细胞性哮喘患者的医生提供了有用的信息。
补充材料
可共享的PDF
确认
作者要感谢以下参与本研究数据收集的研究人员:PR Burgel, C Chenivesse, LJ Couderc, F De Blay, N Freymond, S Fry, PO Girodet, C Leroyer, A Magnan, S Marchand-Adam A Proust, C Raherison-Semjen和N Roche。编辑支持(以写作协助的形式,包括根据作者的指示制定初稿、整理表格和图表、整理作者的评论、语法编辑和引用)由英国Fishawack Indicia有限公司的Sarah Farrar博士和Bianca Paris博士提供,并由GSK资助。
脚注
这篇文章有补充资料可从www.qdcxjkg.com
作者贡献:CT, PC, GD, AD, CP, GG, JC, AB和MH在分析过程中对数据的获取做出了贡献。CT、PC、GD、AD、ChP、GG、AG、CeP、AB、MH对分析概念和设计有贡献;所有作者都对数据的解释、手稿的发展和最终草案的批准做出了贡献。所有作者都可以查阅研究数据。是否发表是作者自己的决定;主办方没有对数据的获取或手稿中的陈述设置任何限制。
利益冲突:C. Taillé报道称,研究和手稿准备支持由葛兰素史克资助;在提交作品之外,葛兰素史克、阿斯利康、诺华、罗氏、赛诺菲、基耶西和TEVA的咨询和讲座的资助和个人费用。
利益冲突:P. Chanez报告说,这项研究和手稿准备支持是由葛兰素史克资助的;Almirall咨询委员会工作的个人费用,Alk-Abello、Boston Scientific和Centocor讲座的个人费用,阿斯利康、勃林格殷格翰、基耶西、GSK、诺华和梯瓦咨询、顾问委员会工作和讲座的个人费用,强生和赛诺菲咨询和顾问委员会工作的个人费用,默克夏普多梅咨询的个人费用,罗氏公司提交工作之外的赠款。
利益冲突:G. Devouassoux报告说,这项研究和手稿准备支持是由葛兰素史克资助的;诺华制药、阿斯利康、葛兰素史克、ALK、TEVA提供的咨询和会议参与资助和个人费用、蒙迪制药提供的咨询和会议参与个人费用、勃林格殷格翰提供的咨询资助和个人费用、Vivisol提供的咨询个人费用、赛诺菲、维塔莱、AB科学、安进、礼来和罗氏提供的资助和参加会议的个人费用、基耶西和默沙东提供的资助和个人费用、AGIR a dom, Orkyn, Stallergene和Takeda参加会议的个人费用,在提交的工作之外。
利益冲突:A. Didier报告说这项研究和手稿准备支持是由葛兰素史克资助的;诺华制药、阿斯利康和葛兰素史克的个人咨询和会议参与费用,奇耶西、勃林格殷格翰、芒迪制药、TEVA、ALK和美纳里尼的个人咨询费用,在提交的工作之外。
利益冲突:C. Pison报告说,研究和手稿准备支持是由葛兰素史克资助的;诺华、阿斯利康、葛兰素史克、基耶西、勃林格殷格翰和赛诺菲为出席会议提供的赠款、个人讲座费用和非经济支持,蒙迪制药和默沙东在提交工作之外为咨询和讲座提供的赠款和个人费用。
利益冲突:G. Garcia报告说,研究和手稿准备支持由葛兰素史克资助;诺华(Novartis)和赛诺菲(Sanofi)咨询委员会工作的个人费用,阿斯利康(AstraZeneca)咨询委员会工作和咨询的个人费用,葛兰素史克(GlaxoSmithKline)咨询的个人费用,Oxyvie在提交工作之外的非经济旅费支持。
利益冲突:J. Charriot报告说,这项研究和手稿准备支持是由葛兰素史克公司资助的。
利益冲突:S. Bouée报道称,该研究和手稿准备支持由葛兰素史克资助;是CEMKA的员工,在提交的工作之外报告来自葛兰素史克的拨款。
利益冲突:A. Gruber报告说,这项研究和手稿准备支持是由葛兰素史克资助的;在提交工作之外,他是葛兰素史克(GlaxoSmithKline)的员工和股东。
利益冲突:C. Pribil报告说,研究和手稿准备支持由葛兰素史克资助;在提交工作之外,他是葛兰素史克(GlaxoSmithKline)的员工和股东。
利益冲突:A. Bourdin报告说,研究和手稿准备支持是由葛兰素史克资助的;来自葛兰素史克、阿斯利康、勃林格殷格翰、基耶西、诺华和赛诺菲的临床试验工作的个人费用,在提交的工作之外。
利益冲突:M. Humbert报告说,研究和手稿准备支持是由葛兰素史克资助的;个人咨询费、会议发言费以及参与阿斯利康、葛兰素史克、诺华、罗氏、赛诺菲和梯瓦的临床研究项目的费用。
支持声明:本研究(GSK ID: 207943;HO-17-18317)由GSK资助。编辑支持由GSK资助。本文的资助信息已存入交叉参考基金注册。
- 收到了2019年12月6日。
- 接受2020年3月14日。
- 版权所有©ERS 2020
本版本根据知识共享署名非商业许可4.0的条款发布。











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}