





文摘
遗传缺陷在骨形态形成蛋白II型受体(BMPRII)信号和炎症导致肺动脉高血压的发病机制(PAH)。受体被激活,骨形成蛋白(BMP)配体,也增强BMPR2转录。小分子与选择性BMP upregulator血管内皮PAH将是一个理想的治疗干预。
我们检测中确定化合物的筛选BMP2 upregulators的能力增加DNA结合的表达抑制剂1 (Id1),使用一个双重驱动的记者专门在人类胚胎干细胞的内皮细胞。这些化验确定了小说哌啶,BMP upregulator 1 (BUR1),增加内皮Id1表达式half-maximal有效浓度为0.098μmol·L−1。微阵列分析和免疫印迹显示BUR1诱导BMP2 prostaglandin-endoperoxide合酶2 (PTGS2)表达式。BUR1有效解救了自体血管生成不足BMPR2+ / R899X内皮细胞生成的CRISPR / Cas9和病人细胞。
野百合碱大鼠BUR1预防和逆转PAH,恢复BMPRII下游信号和调制花生四烯酸途径在肺动脉内皮Sugen 5416 /缺氧PAH小鼠模型。
总之,使用干细胞技术,我们提供了一种新型小分子化合物调节BMP2和PTGS2水平可能有用的治疗多环芳烃。
文摘
干细胞技术确定一种新型小分子化合物调节BMP2和PAH PTGS2水平http://ow.ly/l8R630iqf1R
介绍
肺动脉高压(PAH)是一种进行性疾病由内皮功能障碍和细胞凋亡。它导致肺动脉压力的升高和右心室衰竭(1]。已经提出,不正常内皮细胞受损的信息连接和高渗透性,使促炎的因素渗透到平滑肌层和诱导增殖2]。尽管有大量的多环芳烃疗法的进步,与高死亡率相关的疾病仍然是(3,4]。目前的治疗方法,包括内皮素受体拮抗剂、磷酸二酯酶5抑制剂和环前列腺素类似物,主要影响肺动脉血管收缩(5]。需要额外的方法直接改善血管重塑,正是PAH病理学(6]。
骨形成蛋白(BMP)信号中起着举足轻重的作用在血管系统的发展7)和参与成人脉管系统的维护和体内平衡(8]。杂合的基因突变编码BMP 2型受体(BMPRII)转化的受体增长factor-β总科,被发现在∼80%的家族多环芳烃在15 - 20%的病例和特发性多环芳烃。此外,即使没有可识别的突变,BMPRII表达式在特发性肺血管明显减少多环芳烃(9- - - - - -11]。在一起,这些发现表明,拯救BMPRII毒性较低的信号与小分子监管机构可能是一个容易处理的方法治疗多环芳烃(12]。
维护一个平衡当地生产的endothelial-derived前列腺素类和白细胞三烯在肺血管维持体内平衡是很重要的。Prostaglandin-endoperoxide合酶2 (PTGS2,也称为cyclooxygenase-2)是由缺氧诱导内皮细胞增加前列腺素E2的产量。纯合子PTGS2基因敲除小鼠慢性缺氧下发达国家严重的多环芳烃。相反,花生四烯酸5-lipoxygenase激活蛋白结合花生四烯酸并转移到花生四烯酸5-lipoxygenase (ALOX5),随后去催化成白细胞三烯像白三烯B4(LTB4),高水平的PAH患者已发现和Sugen 5416 /无胸腺的肺动脉高压大鼠模型(13]。此外,ALOX5抑制剂有效减少肺血管重塑在慢性Sugen 5416 /缺氧(苏/重)暴露大鼠(14]。
在这里,我们报告一个人类胚胎干细胞(hESC)中内皮细胞Id1-Venus-Luciferase双记者系统并提供一个小分子BMP upregulator (BUR1)在自体激活BMPRII信号BMPR2+ / R899X人类内皮细胞和病人的细胞通过BMP2。微阵列分析显示,与BUR1 PTGS2也是调节治疗后。在活的有机体内,BUR1不仅预防和逆转PAH的野百合碱(MCT)老鼠还在苏/ Hy鼠模型有影响。
材料和方法
化合物筛选
传统H9为(美国WI Wicell,麦迪逊)稳定转染的构造(Id1-Venus-Luc-MC1-DTA)金星绿色荧光蛋白(GFP)和萤火虫荧光素酶是由完整的人类Id1启动子(±5 kb)。记者细胞被播种在96 -孔板和31个候选化合物被分配到每个(从合成和天然获得医药生物技术研究所的图书馆,中国医学科学院,北京,中国)。4 h后,每个好计算使用的荧光素酶活性Dual-Luciferase®记者分析系统(WI Promega,麦迪逊,美国)。
代R899X杂合的突变
H9的为其定期与30μg co-transfected集群空间短回文的重复序列(CRISPR) / Cas9质粒和30μg E8的捐赠者质粒中过夜通过Lipofectamine®3000,遵循制造商的指示。GFP-positive细胞分类和殖民地的基因组DNA提取。引物序列中列出补充表S1。
苏/ Hy鼠模型
苏/ Hy鼠模型建立了根据先前描述的细节15]。最后六周的模型建立,4.5毫克公斤−1BUR1或车辆控制0.5%的羧甲基纤维素是通过胃内的日常管理(专营)路线三个星期前小鼠接受心肺表现型和被杀。
量化的铂族元素2和LTB4大鼠血浆
老鼠的血液样本收集/沪元模型试验。前列腺素E2(铂族元素2),LTB4使用前列腺素E2水平等离子体测量灵敏度高酶联免疫试剂盒(Abcam,剑桥,英国)和LTB4参数分析工具包(研发系统,明尼阿波利斯,美国)竞争ELISA方法根据制造商的指示。
关于记者的额外信息线代,微阵列,从为内皮细胞分化,CRISPR / Cas9-based突变,免疫染色,未经中华人民共和国交通部,苏/ Hy动物模型和其他实验中可以找到补充材料。
结果
设计和验证的记者hESC细胞系BMPRII信号
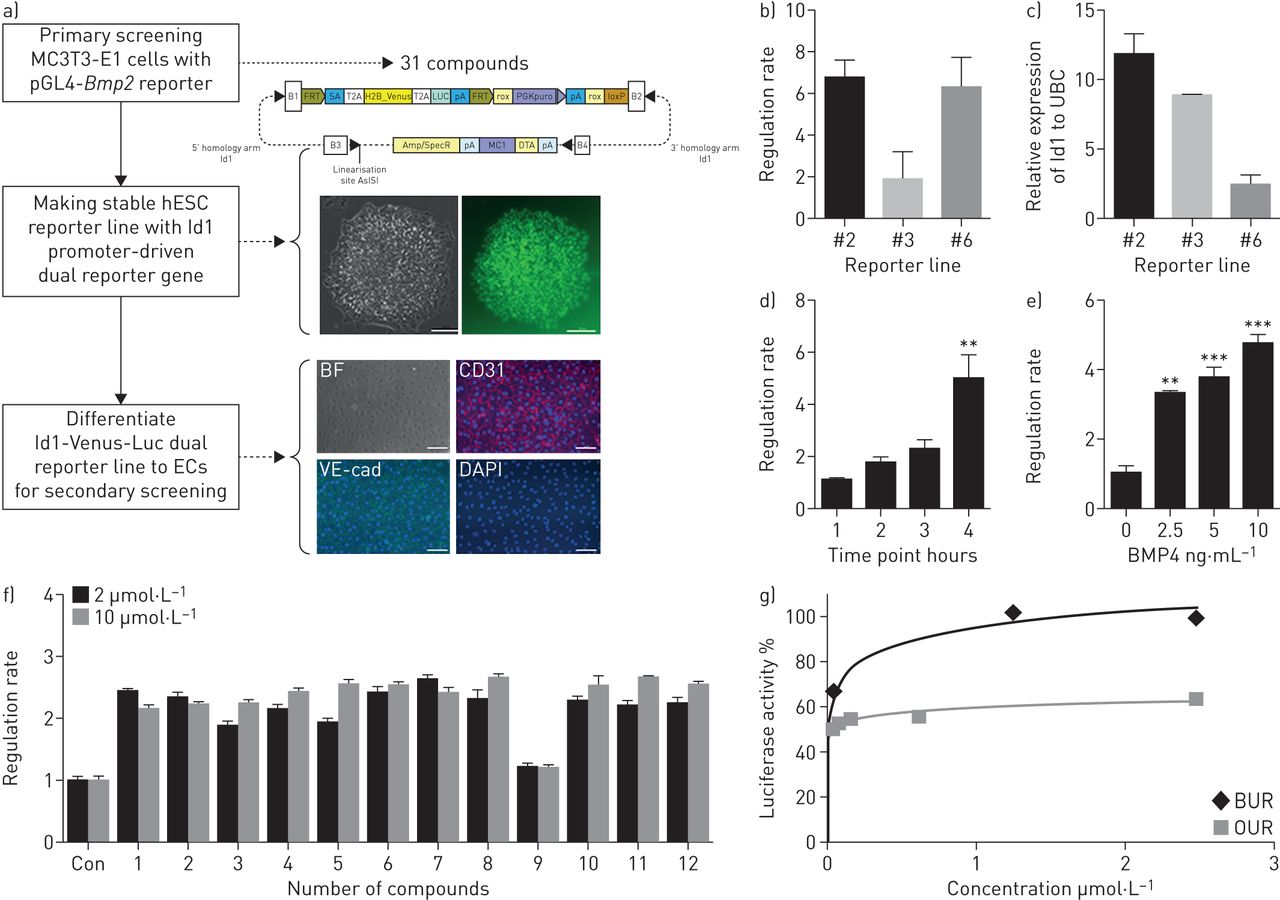
每个位置,包括BMP9和BMP2,增加BMPRII和下游信号在血管细胞(15,16]。小化合物调制BMP9或BMP2临床应用的需要确定。使用BMP2-LUC-MC3T3稳定细胞,31个化合物被确定8160年从先前的筛选小分子化学物质的能力调节BMP2的表达(17]。这些31个化合物组成12的钻头和19 osteoprotegerin(功能)upregulators(我们的)。测试的能力,这些化合物对内皮细胞激活BMPRII信号,我们生成的hESC-Id1-Venus-Luc双重记者行荧光素酶基因全长Id1启动子驱动的进一步验证(图1一个)。虽然克隆2 (CGMCC11091)没有显示最高的活动在BMP4刺激(18,19),这对于验证克隆选择,因为这个细胞系的荧光素酶活性相关的诱导Id1信使rna水平BMP4的存在。这个克隆的BMP4引起的荧光素酶活性表现出时间和浓度模式(图1 b)。证明了Z的因素(0.76),这个细胞系的反应被认为是适合大规模筛选试验(补充表S2)[17]。31个候选化合物中,大多数12车针显示超过2倍荧光素酶活性与车辆控制相比,和达成活动高原低剂量比19的(图1 cd)。
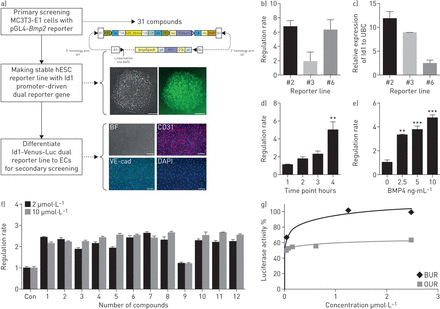
干细胞筛选BMPRII活化剂的信号。一)纲要的主要筛查BMP2 upregulators和一代的双记者人类胚胎干细胞(hESC)派生的内皮细胞(ECs)二级筛查Id1转录的活化剂。看到文本为进一步的细节。代表光明的字段(BF)和荧光图像生成的Id1-Venus-Luc双重记者为(H9)。Id1-Venus-Luc双重记者行分化成ECs;荧光图像显示CD31 VE-cadherin (VE-cad)和DAPI-stained细胞核。酒吧,100μm BF和荧光图像。b) Id1-Venus-Luc活动中检测出三个不同的稳定行4 h后的治疗有或没有10 ng·毫升−1BMP4。c) Id1 mRNA正常化泛素c(哥伦比亚大学)mRNA在相同的三个记者行(n = 3)。d) Id1-Venus-Luc活动确定了在不同时间点(有或没有10 ng·毫升−1BMP4, 1 - 4 h) (n = 4)或e)与不同浓度的BMP4 (0、2.5、5、10 ng·毫升−1)治疗4 h后(n = 4)。f)细胞培养24小时0.1% DMSO(控制(Con))或2μg·毫升−1或10μg·毫升−1从31日候选人化学物质的化合物;相对发光单元被检测到。图显示代表监管利率最钻系列化合物的例子(超过2倍增加控制)(n = 3)。f)百分比代表BUR1和OUR4化合物的荧光素酶活性(n = 3)。* * * * * p < 0.01, p < 0.001。
识别小说BMP2 upregulator人类内皮细胞
进一步验证的有效性钻系列化合物对血管细胞,我们差异化CGMCC11091成CD31-positive细胞(20.)(图1一个和补充的方法)。BUR1 Id1 promoter-driven荧光素酶活性的显著增加CD31-positive内皮细胞摩尔水平。BUR1 (1 - {1-phenyl-1H-pyrazolo [3,4 - d] pyrimidin-4-yl}哌啶)是更有效的比其他11芒刺化合物,显示half-maximal最低有效浓度(EC50)在0.098μmol·L−1。此外,BMPRII被BUR1调节人类肺动脉内皮细胞(PAECs)。因此,它被选为进一步分析(图2一个- c)。最大调节率(荧光素酶活性/控制荧光素酶活性)通过使用1μmol·L−1BUR1 2.23。理解BUR1在内皮细胞的活动,我们使用BUR1刺激血内皮细胞产物(BOECs) PAH患者24小时作为二级筛查和评估使用微阵列分析转录组变化。BUR1诱导微分调节的112个基因(调整假定值0.05),包括BMP2和PTGS2。虽然其他骨形态发生蛋白配体记录与低剂量的细胞被刺激时出现BUR1, BMP2一直调节在所有测试执行(图2 d)。BMP2的增强表达和PTGS2定量实时PCR证实了24 h后BUR1刺激(补充图S1a)。定量实时聚合酶链反应和免疫印迹也证明BMP2的表达和PTGS2 4 h后增加了BUR1刺激(图2 ef)。

识别小说BMP2 upregulator人类内皮细胞。a)荧光素酶活性Id1-Venus-Luc人类胚胎干细胞(hESC)派生的内皮细胞(ECs)刺激后连续稀释浓度的BUR1(0.05 1μmol·L−1)。b) BUR1的化学结构。c)代表免疫印迹显示BMPRII的蛋白表达水平与BUR1刺激后在指定的浓度肺动脉内皮细胞(PAECs)。结果对GAPDH正常化(n = 3)。d)火山块血内皮细胞产物的差异表达基因治疗1μmol·L−1BUR1 (n = 3, p < 0.05,单向方差分析)。e)相对mRNA的表达水平BMP2在PAECs刺激后0.1,1,10 - 25µmol·L−1BUR1介质中含0.1%胎牛血清(的边后卫)4 h;结果正常化GAPDH (n = 3)。代表免疫印迹显示BMP2的蛋白表达水平与BUR1刺激后4 h在PAECs表示低浓度。f)相对mRNA的表达水平PTGS2在刺激后PAECs BUR1为0.1,1,10 - 25µmol·L−1介质中含0.1%的边后卫4 h (n = 3)。代表免疫印迹显示PTGS2的蛋白表达水平与BUR1刺激后4 h在PAECs表示浓度。段落4和6的PAECs应用于这些实验。* * * * * p < 0.01, p < 0.001, * * * * p < 0.0001。电子商务50:half-maximal有效浓度;反对:控制;TGF-β;转化生长因子β;VEGF:血管内皮生长因子。
低剂量的BUR1 Smad1/5加强Id1的表达和磷酸化通过BMPRII PAECs ActRII
我们治疗的细胞与BUR1不同持续时间15分钟至24小时。Smad1/5磷酸化达到30分钟,以最大的刺激和核易位发生1 h;BUR1迅速诱导Id1蛋白质在15分钟,到达了一个高原∼1 h和显示长期表达增加到24 h (图3一,补充图S1)。评估在PAECs BUR1的特异性和有效性,我们治疗细胞与低剂量的BUR1 1 h;BUR1诱导Id1表达PAECs剂量依赖性的方式(图3 b)。浓度为0.2μmol·L−1,BUR1增强Id1和Id2下信使rna表达PAECs(∼2.2倍)。摩尔的BUR1水平增加BMPR2信使rna,这证实了BUR1 BMPRII信号调节的转录水平(图3 c- e,补充图S1a)。考虑到一致性的转录和转译监管BMPRII下游效应器,浓度为0.2μmol·L−1用于以下实验。

低剂量的BUR1加强规范BMP信号通过BMPRII ActRII。)代表免疫印迹和相对光密度分析肺动脉内皮细胞(PAECs)的蛋白表达水平p-Smad1/5和Id1后刺激与0.2μmol·L−1在不同的时间点或1 BUR1 ng·毫升−1BMP9 1 h;结果是GAPDH正常化(n = 3)。b)代表免疫印迹和相对光密度分析PAECs显示Id1的蛋白表达水平和p-Smad1/5刺激与低剂量BUR1(0.01、0.05、0.1或0.2μmol·L−1ng)或1毫升−1BMP9;结果是GAPDH正常化(n = 3)。c)相对mRNA的表达水平BMPR2如果检测到(控制(Con))或与低剂量的刺激在治疗后PAECs BUR1(0.1、0.2或0.5μmol·L−1)1 h。BMP9 (10 ng·毫升−1)是作为一个积极的控制;结果是GAPDH正常化(n = 3)。d, e)相对mRNA的表达水平Id1(d)和Id2下(e)有相同的检测模式与低剂量的治疗后BUR1(0.01、0.05、0.1或0.2μmol·L−1)(n = 3)。f)代表免疫印迹和相对光密度分析PAECs显示p-Smad1/5的蛋白表达水平和Id1刺激后0.2μmol·L−1BUR1 1 h,有或没有pre-incubation与不同浓度(0.01 1μmol·L−1)的激酶抑制剂ldn 30分钟- 193189;结果是GAPDH正常化(n = 3)。g)与单克隆抗体免疫印迹显示的效果BMPRII(BRII)和(或)ActRII(ARII)特殊siRNA Id1蛋白表达;微的结果如下所示(n = 3)。h)与Id1抗体免疫印迹显示个人的影响siRNAs Id1蛋白表达(I型受体)。结果是GAPDH正常化(n = 3)。段落4和6的PAECs应用于这些实验。的边后卫:胎牛血清:NC:新核的控制。* * * p < 0.05, p < 0.01, * * * * * * p < 0.001, p < 0.0001。
进一步调查调解BMPRII信号的激活受体,我们采用选择性抑制剂ldn - 193189块BMP I型受体介导途径。ldn - 193189减毒下游信号剂量依赖性的方式(图3 f)。当我们使用BMP9代替血清,抑制由ldn - 193189还观察到(补充图S2)。接下来,我们证实BUR1和BMP9协同效应的磷酸化Smad1/5 (补充图S3)。只有co-treatment激活素受体蛋白II型(ActRII)和BMPRII siRNA减少Id1表达式到基底的水平(图3 g)。激活素受体1型(ALK2)击倒也减少BUR1-mediated Smad1/5磷酸化(图3 h和补充图S4)。一个截断BMP2启动子分析表明Smad绑定元素,而不是视黄酸反应元素,负责BMP2转录激活BUR1刺激(补充图S5)。我们还测试了其他类型的细胞,包括BOECs人类脐静脉内皮细胞、心肌细胞和人类肺动脉平滑肌细胞。BMPRII的表达增加了BUR1 BOECs和人类脐静脉内皮细胞,但不是在心肌细胞。肺动脉平滑肌细胞显示轻度BMP2蛋白表达增加,心肌细胞一样,和一个小BMPRII信号相比,内皮细胞的变化。PTGS2也提升了BUR1治疗心肌细胞(补充图S6)。总的来说,这些发现表明,BUR1上调BMP2通过Smad绑定元素,然后提高BMPRII信号通过ALK2主要在内皮细胞。
BUR1提高规范化和非规范BMP信号和逆转管形成缺陷BMPRII-deficient内皮细胞
测试BUR1对遗传的影响多环芳烃(HPAH)引起的过早停止突变,我们采用CRISPR / Cas9系统生成R899X自体杂合的突变(图4一- d,补充图S7,补充表S3和S4)。评估haploinsufficiency对血管生成的影响,我们区分目标与自体野生型为并行线。配体的表达组织Apelin,特别结合G protein-coupled受体已在内皮细胞(21),显著降低了杂合的细胞系(图4一- c) [22]。我们使用有区别BMPR2+ / R899X内皮细胞评估BMPRII信号BUR1,我们观察到增强组织Apelin Id1表达式(图4 d)。利用从患者BOECs特发性PAH,我们也证明了BUR1增强BMPRII信号(图4 e)。此外,BUR1获救管网络的形成同样在患者内皮细胞血管内皮生长因子(图4 f我),BMPR2+ / R899X内皮细胞(补充图S7d)。尽管Id1众所周知,提高低氧诱导因子(HIF)蛋白质的稳定促进管形成,我们发现HIF-1αHIF-2α及其下游目标arginase-1没有显著影响培养PAECs或肺动脉的苏/ Hy-treated老鼠(补充图S8)。

在自体BUR1增强BMP信号和管形成BMPR2+ / R899X内皮细胞(ECs)和血内皮细胞产物(BOECs)患者肺动脉高血压(PAH)。一)形态变化和相对mRNA的表达BMPR2在EC感应在第0天,第五天,第十天BMPR2野生型(Wt)和杂合的(BMPR2+ /R899X,叫)细胞系。酒吧,规模200μm。b)免疫印迹和c)定量实时PCR结果显示BMPRII的蛋白质含量和mRNA水平和组织Apelin Wt叫ECs,分别为(n = 3)。d)蛋白表达水平的组织Apelin和Id1 Wt,叫ECs。结果是GAPDH正常化。细胞如果(控制(Con))或刺激1 ng·毫升−1他克莫司(吸收FK506)或0.2或1μmol·L−1BUR1 0.1%血清1 h serum-starvation后16 h (n = 3)。e)定量实时聚合酶链反应和免疫印迹结果表明的mRNA水平BMPR2和蛋白质水平的BMPRII, p-Smad1/5和Id1 BOECs来自健康供者(HD)和PAH患者GAPDH正常化。细胞如果(Con)或刺激0.2μmol·L−1BUR1(定量实时PCR)或0.2或1μmol·L−1钻(免疫印迹)0.1%的血清饥饿后4 h (n = 3)。f)代表图像BOECs高清或管形成的多环芳烃在各种条件。serum-starvation 16 h后,总共20 000个细胞与基底膜基质添加到48-well板板。这些细胞被刺激30 ng·毫升−1血管内皮生长因子(VEGF)或0.2μmol·L−1BUR1并行,如果细胞控制(Con)。管状结构的形成是发现4 h种子期后。酒吧,规模200μm。g连接的数量,h)数量的分支和我)分支机构的总长度在10每个试验定量分析领域使用ImageJ软件(n = 3)。段落4和6的BOECs应用于这些实验。黑色箭头表示16 kDa的二聚体形式组织Apelin蛋白质。* * * p < 0.05, p < 0.01, * * * * * * p < 0.001, p < 0.0001与反对在相应的组;# #p < 0.01,# # #p < 0.001,# # # #p < 0.0001与高清的。
BUR1阻止和逆转MCT-induced PAH的老鼠
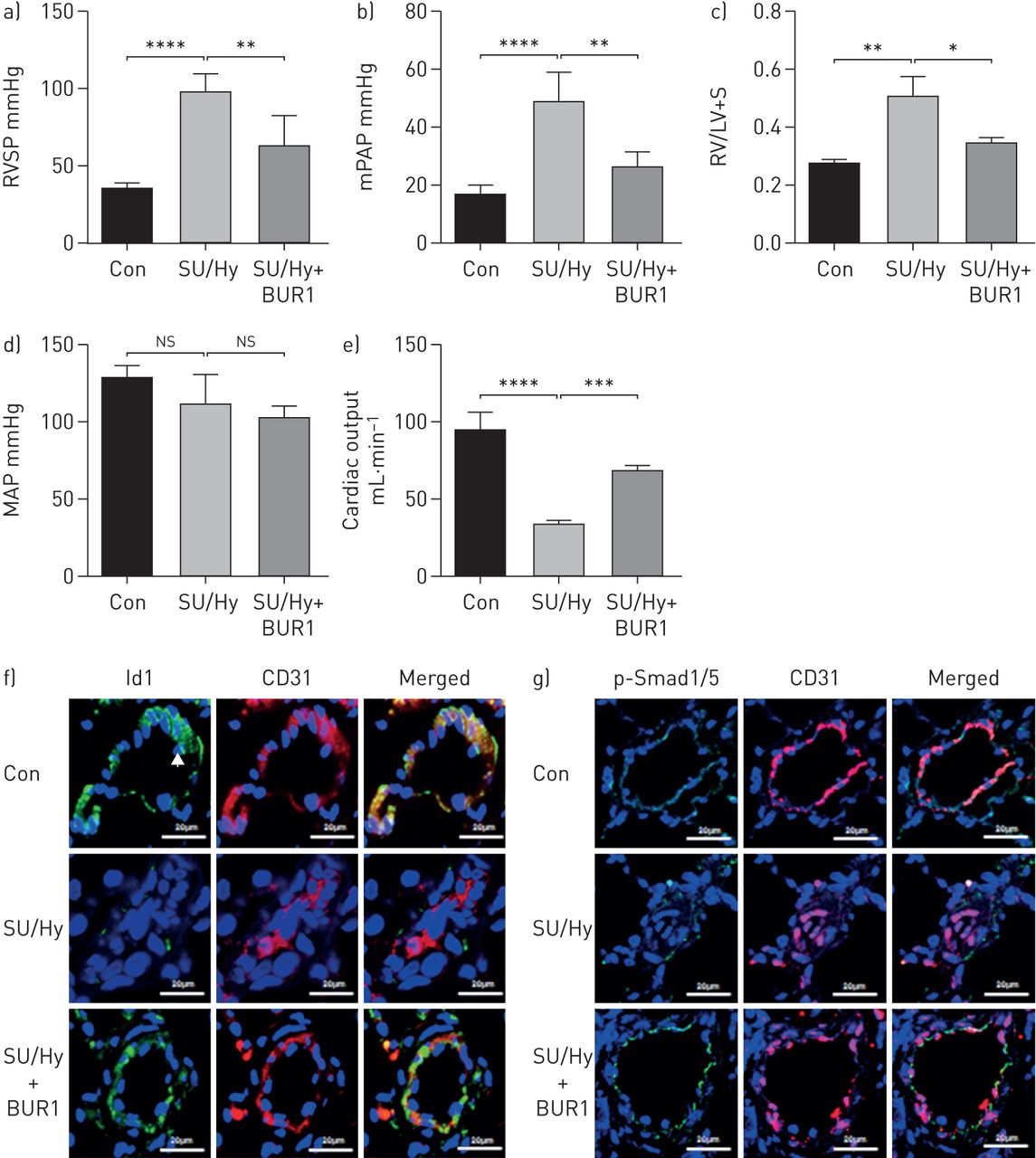
接下来,我们评估是否BUR1可以防止血管重塑PAH动物模型(23]。所示图5一个- d, BUR1治疗3周(4.5 mg·−1,专营)发起1星期后MCT注射导致显著降低肺动脉压力和右心室收缩压(RVSP 35±1.5 mmHg)相比saline-treated组(45±1.5毫米汞柱)。治疗BUR1也阻止了右心室肥大和增厚的外周肺动脉MCT老鼠。虽然预防性治疗与其他抗高血压药,芒柄花黄素(24],减少RVSP MCT-PAH模型中,非特异性降低平均系统性动脉压力(MAP)排除它作为一个潜在的治疗多环芳烃。比较日常管理的有效性的0.45、4.5和45毫克公斤−1老鼠(专营)透露,中、高剂量的BUR1有相似的动物模型中对疾病预防和生存的影响没有系统性血压的变化(图5一个- d)。
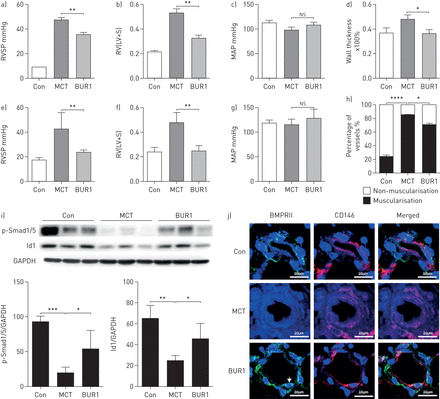
全身的BUR1阻止和逆转野百合碱(MCT)肺动脉高血压。)评估右心室收缩压(RVSP), b)右心室肥大(右心室(RV)重量比左心室(LV) +隔(S)重量(RV / (LV + S))和c)平均动脉压(MAP)在大鼠在预防协议给定车辆(控制(Con), n = 6)或MCT(皮下注射(南))为1周,随后处理车辆(MCT, n = 6)或BUR1 (BUR1,胃内的(渐开线齿轮),n = 6)从8到21天。d)肺动脉壁厚被媒体壁厚的比例量化直径(直径50 - 100μm, n = 6为每个组,10个字段为每个幻灯片)。e)评估RVSP f) RV / (LV + S)和g)地图的老鼠在逆转协议给定车辆(Con)或MCT(55毫克公斤−1,南卡罗来纳州。,n=18) and subsequently treated with saline (MCT, n=9) or BUR1 (BUR1, 4.5 mg·kg−1n = 9)从天21-35 post-MCT;分析了h) muscularisation与直径30 - 80μm肺动脉。我)代表免疫印迹和密度测量分析的蛋白表达水平的磷酸化Smad1/5和Id1整个肺溶解产物从老鼠发生逆转治疗(n = 3)。j)代表共聚焦免疫荧光染色的图片5张幻灯片BMPRII(绿色)和肺的CD146(红色)部分的控制(Con)和MCT-exposed老鼠用生理盐水治疗(MCT)或BUR1 (BUR1)逆转后协议(n = 6)。箭头表示肺动脉内皮细胞。核与DAPI复染色(蓝色)。酒吧、规模20μm。ns,不重要。* * * p < 0.05, p < 0.01, * * * p < 0.001。
管理BUR1 2周后21天的接触MCT证实的疗效BUR1治疗逆转MCT-induced损伤(图5 e- h)。显著降低RVSP和右心室肥大被记录了下来。没有明显差异在地图或体重MCT盐水——和BUR1-treated MCT老鼠(数据未显示)。外围的muscularisation肺动脉也部分逆转了BUR1治疗(图5 h)。免疫印迹和量化p-Smad1/5 Id1从控制在整个肺细胞溶解产物,MCT盐水——和BUR1-treated MCT老鼠显示规范BMP信号提高BUR1-treated组(图5我)。BMPRII cd146内皮细胞染色检测;的表达下调BMPRII表达式MCT老鼠被BUR1救起治疗(图5 j)。改进BMPRII下游信号、防扩散和巨噬细胞浸润减少造成BUR1治疗进一步证实了免疫染色的肺动脉BUR1-treated老鼠(补充图S9)。评估慢性治疗后碱性磷酸表达式,使用BMP2积极控制,表明BUR1没有对C2C12细胞(成骨的影响补充图S10)进一步药理研究表明,BUR1低毒性和半衰期约为12.7 h在水溶液37°C (补充图S11和S12)。
BUR1变弱多环芳烃在苏/ Hy鼠模型提高BMP信号
单一剂量的Sugen管理后,老鼠被放置在3周的慢性缺氧条件,其次是抓捕normoxia的时期。最后normoxia时期,苏/ Hy-treated老鼠显示增加RVSP(97±12毫米汞柱)与对照组相比(34±5毫米汞柱)。在接下来的3周,4.5毫克公斤−1BUR1的日常管理。BUR1逆转了PAH,包括减少RVSP,意味着肺动脉高血压和右心室肥大(图6- c)。没有明显的差异之间的映射苏/和苏/ + BUR1老鼠为什么(为什么图6 d)。左心室功能评估表明,心输出量和三尖瓣环平面收缩偏差(TAPSE)改善苏/老鼠为什么对待BUR1相比,车辆(图6 e,补充图S13和表S5)。我们还发现,p-Smad1/5 Id1信号与CD31-positive co-localised肺动脉内皮细胞远端动脉。苏/老鼠为什么Id1 p-Smad1/5染色了肺血管,减少由apoptosis-resistant内皮前体细胞。最后,调节规范BMP信号确认BUR1-treated组(图6 fg)。
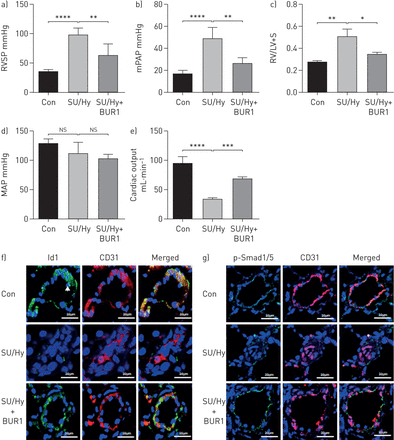
BUR1变弱Sugen 5416 /缺氧(苏/重)全身肺动脉高血压。老鼠给车辆(控制”(体质)、“n = 6)或挑战su - 5416(皮下(南))和维护在缺氧3周然后normoxia为3周。每日治疗后与羧甲基纤维素车辆(苏/ Hy, n = 6)或BUR1(苏/ Hy + BUR1,胃内的(渐开线齿轮),n = 6) 3周,血流动力学指标的)右心室收缩压(RVSP), b)的意思是肺动脉高血压(肺动脉平均)和c) RV / (LV + S) + d)平均动脉压(MAP)和e)心输出量是9周的老鼠。f, g)代表共焦图像的免疫荧光染色从五个幻灯片Id1(绿色)和CD31(红色)(f)和CD31(红色)和p-Smad1/5(绿色)(g)从控制、苏/衔接和苏/ Hy + BUR1肺部分(n = 6)。核与DAPI复染色(蓝色)。白色箭头表示Id1的定位在细胞核中。酒吧、规模20μm。ns,不重要。* * * p < 0.05, p < 0.01, * * * * * * p < 0.001, p < 0.0001。
BUR1调节PTGS2和花生四烯酸5-lipoxygenase肺动脉内皮细胞激活蛋白
PTGS2参与BUR1-attenuated多环芳烃可能成为另一个重要因素。花生四烯酸代谢途径的另一个重要调节器,花生四烯酸5-lipoxygenase激活蛋白(ALOX5AP),显示一个相反的表达趋势当BUR1处理(图7)。长期与BUR1孵化24小时持续改变PTGS2 ALOX5AP蛋白质水平的记录在更早的时间点(图2 f和图7 b)。的表达Ptgs2和苏Alox5ap增加/ Hy鼠肺溶解产物(图7 c苏,d)和增厚脉管系统的/ Hy鼠肺。Ptgs2表达维持在高水平在苏/ Hy集团,而Alox5ap的表达减少到基线水平的治疗BUR1 (图7 e,f)的表达PTGIS和TBXAS1下游PTGS2酶的级联,不同受BUR1 PAECs刺激(补充图S14a和S14b)。此外,基因的表达参与了LTB4代谢明显降低肺的BUR1-treated动物相比苏/ Hy老鼠(补充图S14c)。进一步了解综合调制类二十烷酸代谢物,我们测量铂族元素2和LTB4等离子体水平的苏/ Hy老鼠和那些BUR1处理。虽然都是调节在苏/ Hy鼠血浆,LTB4演示了一个健壮的减少BUR1治疗(图7 g,h)。综上所述,这些结果表明,BUR1调制PTGS2对肺血管的抗炎效果。

BUR1调节PTGS2和ALOX5AP肺动脉内皮细胞(PAECs)和大鼠肺。一)相对mRNA和蛋白表达的ALOX5AP PAECs BUR1 4 h与不同剂量治疗后;蛋白表达的光密度分析显示在右面板(n = 3)。b)代表免疫印迹显示PTGS2的表达和ALOX5AP PAECs治疗24小时后用不同剂量的BUR1;光密度分析显示在右面板(n = 3)。c, d)相对mRNA的表达Ptgs2(c)和Alox5ap(d)在整个肺溶解产物从老鼠(n = 3)。e、f)代表图像的免疫荧光染色从五个幻灯片Ptgs2 (f,红色)或Alox5ap (g,红色)和CD31(绿色)从控制、苏/衔接和苏/ Hy + BUR1肺部分(n = 6)。核与DAPI复染色(蓝色)。酒吧、规模20μm。g)铂族元素2和h) LTB4等离子体的水平苏/苏Hy老鼠和老鼠为什么对待BUR1 (n = 3)。段落4和6的PAECs应用于这些实验。ns:不重要。* * * * p < 0.05, p < 0.0001。
讨论
之前的报告建议增加内皮BMPRII信号作为一个策略来提高多环芳烃的肺血管重塑。在这里,我们报告一个新的BMP2监管机构,控制BMPRII BUR1 PAECs信号和花生四烯酸途径。它有效地防止和逆转疾病的多环芳烃在老鼠模型。提供一个自体和类似的遗传模型PAH,我们还生成一个致病突变hESC-derived内皮细胞评估新的小分子化合物的作用机制。
BMP信号是由heteromeric受体复合物的形成II型和I型受体(25]。BMPRII信号包括规范化和非规范途径,都是受到多环芳烃BMPRII缺陷的影响。规范BMP信号包含Id蛋白质,作为下游目标Smad1/5/8激活(26- - - - - -28]。Id1转录显著增加了BMP2内皮细胞;因此,我们结合一个初级筛查upregulator BMP2的二级筛查Id基因转录的激活hESC-derived内皮细胞选择性地缩小范围的积极。我们双记者行分化成内皮细胞来测试化合物选择性。
BUR1 pyrazolopyrimidine核心具有平面结构。最近的报告结构的{1 h-pyrazolo [3,4 - d] pyrimidin-4-yl}哌啶化合物,哪些功能serine-threonine和酪氨酸激酶调制器治疗免疫,炎症和增生性疾病。BUR1 EC最低50upregulation检查类似物的BMP2和Id1转录。我们认为BUR1是否有相同的动作pyrimidinone西地那非的核心,而竞争性块环鸟苷酸的水解5型磷酸二酯酶(PDE5)。然而,尽管这些化合物结构相似,BUR1 PDE5A1演示了一个低得多的抑制作用,显示值抑制浓度(IC50)> 20μmol·L−1。因此,它是可能的,BUR1行为通过酪氨酸激酶调节BMP信号。我们也调查了脱靶效应BUR1 C2C12细胞的骨生成和显示BUR1不能产生碱性磷酸酶活动BMP2一样的程度。
BUR1演示了其潜在的监管机构BMPRII信号EC500.098μmol·L−1在hESCs-Id1-Venus-Luc记者系统。此外,BUR1增强BMP2和PTGS2表达内皮细胞。我们还发现,低剂量BUR1有效增加Smad1/5激活和PAECs Id1转录。连续的药物发现从标准筛选微摩尔的水平的选择性和有效剂量BUR1(0.2μmol·L−1)。在早期的时间点,BUR1影响BMPRII信号。在后来的时间点,间接影响PTGS2和其他因素日益明显,这可能是有益的长期治疗。我们发现BMP2一直调节从早期的时间点到24 h,当传达信号增强BMPRII的表达。我们进一步调查的机制BUR1激活BMPRII信号在早期时间点通过ldn - 193189块I型和II受体0.1%血清的存在。我们发现规范BMP信号是剂量依赖性的方式减少。我们的发现表明存在剂量依赖的相关性的封锁ldn - 193189在BUR1激活BMP9的存在。我们用专门设计的siRNA击倒BMPRII ActRII受体,和减少Id1涉及这两种受体在BUR1-stimulated组件规范BMP信号。启动子缺失分析进一步澄清,BUR1调节BMP2转录增强Smads绑定在其启动子。
我们使用CRISPR / Cas9基因定位工具来生成R899X为点突变。该模型系统有许多好处。首先,该系统提供了一个无限的,可扩展的资源的人类BMPR2+ / R899X内皮细胞。尽管嘌呤霉素完全恢复从获得的突变细胞中转录BMPR2+ / R899X鼠标,它表现出在人类细胞(只有部分的效果15]。其次,该系统生成一个自体对应变异减少生物个体之间的变异。最后,它可以研究遗传因素的影响对人类的发展主题。最近的一项研究表明,诱导多能干细胞诱导/遗传PAH分化成内皮细胞有相同的基因表达谱PAECs从同一个病人29日]。剩下的BMPRII保持基底激活规范BMP信号通过Smads BUR1。我们确认海拔BMPRII信号使用相同的治疗内皮细胞从PAH患者BMPRII不足。
我们发现PTGS2表达明显增加,同时减少ALOX5AP表达BUR1-treated内皮细胞和苏/ Hy鼠肺动脉。我们还发现PTGS2表达增加可能是通过在PAECs BMP2 BUR1治疗引起的。BMP2报道激活激活转录因子2和runt-related近端启动子的转录因子2绑定到PTGS2 [30.]。与此同时,减少了LTB BUR1治疗4生产,由于减少了表达ALOX5AP导致花生四烯酸的氧化。我们还发现一个轻微的铂族元素的增加2生产的血管舒张药血浆从对待苏/ Hy老鼠。铂族元素2是一个关键的中介炎症诱发pro -抗炎作用和信号通过四种不同signalling-E前列腺素类(EP)受体,EP1 EP4 (31日,32]。铂族元素2展品中的各种影响肺,肺血管重塑和炎症等监管(33]。在这项研究中,花生四烯酸代谢的平衡在PAECs被BUR1将有利于PTGS2而不是ALOX5AP活动,导致高架铂族元素2/ LTB4比例从BUR10-treated肺动脉高压大鼠肺动脉内皮细胞。这些结果表明,BUR1调节PTGS2有利于抗炎反应实验PAH (图8)。
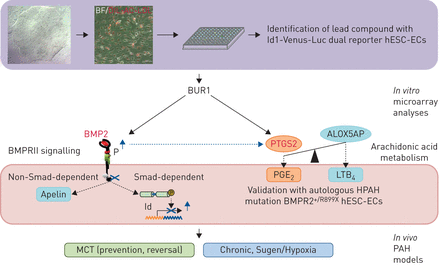
提出了干细胞药物筛选和验证系统。BUR1,哌啶,Id1-Venus-Luc双重记者确认了人类胚胎干细胞(hESC)中内皮细胞(ECs)作为BMPRII信号调节器从31个小分子化合物。在体外,BUR1增强BMP2的转录,之后,在肺动脉PTGS2 ECs。其效果验证的自体遗传肺动脉高血压(HPAH)突变BMPR2+ / R899XhESC-ECs和特发性PAH血液ECs产物。Smad-dependent通路(Id)和non-Smad-dependent通路(如。组织Apelin)被激活,促进血管生成BMPRII-deficient ECs。BUR1疗效的多环芳烃进一步验证的野百合碱(MCT)多环芳烃鼠模型和Sugen 5416 /缺氧(苏/重)多环芳烃大鼠模型在活的有机体内。这项工作表明干细胞药物筛选的新系统。
我们的在活的有机体内研究表明潜在的PAH BUR1作为治疗剂。我们测试了三种不同剂量的BUR1预防研究中使用特定的老鼠模型,选择4.5毫克的剂量·公斤−1·天−1,这被证明是有效的多环芳烃在阻止病情发展。建立了多环芳烃的逆转MCT老鼠通过与BUR1治疗,减少管理中华人民共和国交通部负责RVSP和内侧肥大。我们还演示了恢复BMPRII信号BUR1-treated肺动脉壁的老鼠。本研究的更有前途的结果是获得研究慢性缺氧时一起Sugen PAH的诱导大鼠模型,血液动力学的测试后,包括超声心动图;在这个实验中,BUR1不仅减毒磷酸化Smad1/5 Id1蛋白质含量,而且监管的平衡PTGS2和ALOX5AP表达血管内膜损伤,这表明针对BMPRII信号加强抗炎效应多环芳烃是一个有效的治疗策略。
总之,这项研究提供了基础治疗的发展战略目标BMPRII信号的多环芳烃。使用干细胞记者行中,我们发现了一个选择性BMP2 upregulator内皮细胞。增强BMPRII信号通过观察BUR1不仅在PAECs还在自体内皮细胞和变异hESC BOECs获得PAH患者。BUR1也将花生四烯酸代谢PTGS2-driven抗炎作用。使用未经中华人民共和国交通部和苏/ Hy PAH动物模型允许我们提供一个新颖的结构类型多环芳烃和其他相关疾病的治疗(图8)。
补充材料
确认
我们要感谢Mariaestela奥尔蒂斯(Wellcome Trust-MRC剑桥大学干细胞研究所,剑桥,英国)设计双重记者陈线和仲秋节(基础医学科学研究所、中国医学科学院、北京协和医学院、北京、中国)进行启动子部分删除结构。我们还要感谢肖建王(心血管疾病国家重点实验室、阜外医院、北京、中国)帮助动物实验。
脚注
可以从本文的补充材料www.qdcxjkg.com
作者的贡献:j .杨设计研究和完成手稿;y兴进行了动物实验和PTGS2-related研究和修订后的手稿;问:魏、赵,f .周和r . Al-Lamki进行实验并获得数据;g .商执行化学筛选;赵x进行免疫印迹实验;d . Ortmann和m . Du进行了干细胞的实验;r·皮德森提供干细胞相关试剂;美国如果提供化学物质;西北Morell修订后的手稿并提供细胞培养试剂。
支持声明:这项工作是国家重点支持的研究和发展项目的中国——干细胞与转化研究(编号2016 yfa0102300),凸轮医学科学创新基金(CIFMS 2016 - i2m 4 - 003),中国国家自然科学基金(国家自然科学基金委)(81400278和81400278),中国千(年轻)人才项目和基础研究基金中央大学j·杨;国家自然科学基金委s . Si (81621064);和y兴国际博士后交流奖学金计划。资金信息,本文已沉积的Crossref资助者注册表。
利益冲突:没有宣布。
- 收到了2017年10月31日。
- 接受2018年2月10日。
- 版权©2018人队











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}