





Abstract
We evaluated the efficacy and safety of ciprofloxacin dry powder for inhalation (DPI) in patients with non-cystic fibrosis bronchiectasis, two or more exacerbations in the previous year and predefined sputum bacteria.
Patients were randomised 2:1 to twice-daily ciprofloxacin DPI 32.5 mg or placebo in 14- or 28-day on/off treatment cycles for 48 weeks. Primary end-points were time to first exacerbation and frequency of exacerbations. Enrolling countries and α level split (0.049 and 0.001 for 14- and 28-day cycles, respectively) differed from RESPIRE 1.
Patients were randomised to ciprofloxacin DPI (14 days on/off (n=176) or 28 days on/off (n=171)) or placebo (14 days on/off (n=88) or 28 days on/off (n=86)). The exacerbation rate was low across treatment arms (mean±SD.0.6±0.9). Active treatment showed trends to prolonged time to first exacerbation (ciprofloxacin DPI 14 days on/off: hazard ratio 0.87, 95.1% CI 0.62–1.21; p=0.3965; ciprofloxacin DPI 28 days on/off: hazard ratio 0.71, 99.9% CI 0.39–1.27; p=0.0511) and reduced frequency of exacerbations (ciprofloxacin DPI 14 days on/off: incidence rate ratio 0.83, 95.1% CI 0.59–1.17; p=0.2862; ciprofloxacin DPI 28 days on/off: incidence rate ratio 0.55, 99.9% CI 0.30–1.02; p=0.0014), although neither achieved statistical significance. Ciprofloxacin DPI was well tolerated.
Trends towards clinical benefit were seen with ciprofloxacin DPI, but primary end-points were not met.
Abstract
RESPIRE 2 supports ciprofloxacin DPI clinical benefits in bronchiectasis, but did not reach statistical significancehttp://wly/es9e30gnhos.
Introduction
Non-cystic fibrosis bronchiectasis (NCFB) is a heterogeneous disease with a range of underlying aetiologies across geographical regions and a variable clinical course [1]。Patients with NCFB are subject to a vicious cycle of inflammation, poor mucus clearance and recurrent infection [2,3]。这种循环表现为慢性咳嗽,痰液生产,气道损伤和加剧,每次加剧的严重程度和影响都会从患者到患者,并对进步肺部损伤负责[2,4]。
支气管扩张的恶化是症状的衰弱增加,与较差的健康结果相关,包括减少生活质量,抑郁,焦虑和死亡率增加[5–7]。因此,降低加剧的频率是一个重要的治疗目标;但是,目前没有许可抗生素治疗。
t的呼吸计划包括两个试验he same overall design, as required by regulatory agencies. The objective of both studies was to assess the efficacy and safety of 14- and 28-day on/off regimens of ciprofloxacin dry powder for inhalation (DPI) in prolonging time to first exacerbation and reducing frequency of exacerbations in patients with NCFB. The RESPIRE 1 trial demonstrated that treatment with a 14-day on/off cycle of ciprofloxacin DPI significantly reduced frequency of exacerbations and prolonged time to first exacerbation in patients with NCFB. The 28-day ciprofloxacin DPI arm of RESPIRE 1 had trends towards clinical benefit, but failed to reach statistical significance [8]。Here, we report the results of the phase III RESPIRE 2 trial (ClinicalTrials.govidentifierNCT02106832)。呼吸1和2之间的主要差异是招聘国家的变化(呼吸2吸引更多来自亚洲和东欧的患者,而不是呼吸1),以及对统计计划的具体变更,如以下部分所述。
方法
Study design and treatment
The RESPIRE trials had the same overall methodology, which has been reported previously [9]。Two interventions were studied over 48 weeks: ciprofloxacin DPI 32.5 mg twice daily 14 days on/off (12 cycles) and ciprofloxacin DPI 32.5 mg twice daily 28 days on/off (6 cycles). There was an 8-week off-treatment follow-up period.
Study population
患有初步诊断NCFB(特发性或发作性或感染性疾病)和稳定疾病的患者。关键纳入标准是:前12个月至少两次恶化,并在筛选七种预定病原体中的至少一种筛选阳性痰培养物(Pseudomonas aeruginosa,嗜血杆菌流感,Moraxella catarrhalis,Staphylococcus aureus,Streptococcus pneumoniae,Stenotrophomonas maltophiliaorBurkholderia cepacia)。Patients with active allergic bronchopulmonary aspergillosis, active or actively treated nontuberculous mycobacterial lung infection or tuberculosis, a primary diagnosis of chronic obstructive pulmonary disease (COPD), or documented chronic asthma were excluded. Randomisation was stratified by geographical region, pre-therapy positive culture forP.铜绿假单胞菌and chronic macrolide use.
终点
Two different hierarchical statistical analysis approaches were developed with different regulatory authorities. The primary end-point for the US Food and Drug Administration (FDA) was time to first exacerbation within 48 weeks after start of treatment (ciprofloxacin DPI与合并的安慰剂)。对于欧洲药物局(EMA)/其他机构,如中国国家药物管理局,初级终点是在48周的研究期间发生恶化的频率(CiProfloxacin DPI与匹配的安慰剂)。汇集安慰剂组引用to all patients from the 14- and 28-day on/off placebo arms; matching placebo refers to ciprofloxacin DPI 14 days on/off compared with placebo 14 days on/off and ciprofloxacin DPI 28 days on/off compared with placebo 28 days on/off [4]。For the primary end-points, exacerbations had to meet three criteria: 1) worsening of at least three signs or symptoms (dyspnoea, wheezing, cough, 24-h sputum volume or sputum purulence) beyond normal day-to-day variations for at least 2 consecutive days, 2) fever (body temperature >38.0°C) or malaise/fatigue and 3) systemic antibiotic treatment (“primary end-point definition”). Pre-defined subgroups and safety end-points are described in补充部分S1和S2)。次要终点have been detailed previously [8,9]。A secondary end-point of particular interest was exacerbation rate according to a less stringent definition,i.e.一种呼吸事件,具有恶化至少一个上述症状或症状和全身抗生素使用。
Analyses
The statistical analysis plan for the RESPIRE programme has been described previously [9]。呼吸2跟随与呼吸1相同的分析计划,有两个例外。首先,在FDA和EMA /其他关于处理中断的处理之间分析FDA和EMA /其他分析的频率分析模型。在呼吸1 FDA分析中,对于未完成48周的患者,外推的频率外推,而在呼吸2中,患者在FDA分析中将患者的研究时间作为EMA /其他分析作为偏移变量。。其次,在研究结束之前修改了显着性水平(α水平分裂),基于从呼吸1.在呼吸1中,使用Bonferroni校正,导致每个治疗臂的显着性水平为0.025(14和28天开关)。在呼吸1的CiProfloxacin DPI 28天开/关臂的统计失效之后,呼吸2的α校正被前瞻性地修正为0.049的加权Bonferroni校正,为14天开/关臂和0.001的28-一天开/臂。赞助公司在学习结束之前讨论了这一变化,并批准了这一变革的监管机构。进一步的细节是给出的补充部分S3。
Results
Patients
A total of 521 patients from 24 countries were randomised to treatment (figure 1); the first patient visit was in April 2014 and the last visit was in October 2016. The highest enrolling countries were Russia (11.5%), Bulgaria (10.4%) and Latvia (8.6%) (补充表S1)。
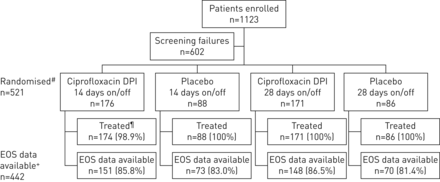
Patient disposition. DPI: dry powder for inhalation; EOS: end of study.#:由地理区域分层的随机化,预治疗阳性文化Pseudomonas aeruginosaand chronic macrolide use;¶:患者随机接受至少一种治疗剂量;+:EOS数据的患者包括完成治疗和随后的后续的患者,或过早停止治疗,但遵循计划的EOS(52/54周),或过早停止治疗,但完成了计划的8周后的计划。
患者人口统计数据和基线特征在群体中平衡(table 1)。The majority of patients (58.0%) were female, the mean age was ∼60 years and patients had a high likelihood ofP.铜绿假单胞菌infection (60.7% of patients; see补充表S2for additional pre-specified baseline pathogens). Lung function was generally poor, with >40% of patients having a forced expiratory volume in 1 s (FEV1) % pred <50%. Approximately 30% of patients had a medical history of COPD and use of concomitant respiratory medications considered part of NCFB therapy was common (补充表S3)。
Patient demographics and baseline characteristics
44.2 patients (84.8%) completed the study and 79.5% of patients (414 out of 521) completed treatment. Patient compliance was high (mean >95% in all arms) (table 2)。
Treatment compliance and duration#
主要终点
Both the 14- and 28-day ciprofloxacin DPI on/off arms increased the time to first exacerbation与汇集安慰剂(figure 2)。However, neither ciprofloxacin DPI treatment arm demonstrated a statistically significant difference compared with pooled placebo (ciprofloxacin DPI 14 days on/off: hazard ratio (HR) 0.87, 95.1% CI 0.62–1.21; p=0.3965 and ciprofloxacin DPI 28 days on/off: HR 0.71, 99.9% CI 0.39–1.27; p=0.0511) (table 3)。The median time to first exacerbation was not estimable for any treatment arm due to the low exacerbation rates. Overall, 61.4% and 67.3% of patients treated with ciprofloxacin DPI 14 and 28 days on/off, respectively, did not experience an exacerbation matching the stringent definition, compared with 58.0% of patients in the pooled placebo arm.
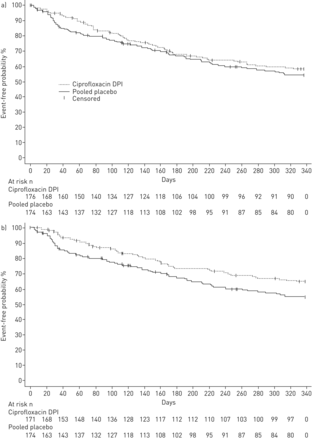
Time to first exacerbation for patients receiving ciprofloxacin dry powder for inhalation (DPI) a) 14 days on/off or placebo and b) 28 days on/off or placebo. For the primary end-point exacerbations were required to meet three criteria: 1) worsening in at least three signs or symptoms (dyspnoea, wheezing, cough, 24-h sputum volume or sputum purulence) beyond normal day-to-day variation for at least 2 consecutive days, 2) fever (body temperature >38.0°C) or malaise/fatigue, and 3) systemic antibiotic treatment.
主要终点: time to first exacerbation and frequency of exacerbations#在用环丙沙星干粉治疗的患者中吸入(DPI)与placebo over 48 weeks
Ciprofloxacin DPI 14 days on/off reduced the exacerbation incidence rate by 17% over 48 weeks (incidence rate ratio (IRR) 0.83, 95.1% CI 0.59–1.17; p=0.2862), while ciprofloxacin DPI 28 days on/off reduced the exacerbation incidence rate by 45% (IRR 0.55, 99.9% CI 0.30–1.02; p=0.0014) (table 3)。Neither arm reached the predefined statistical significance level (p=0.049 for the 14-day regimen and 0.001 for the 28-day regimen). Overall, the frequency of exacerbations in all arms was very low (figure 3)。总体平均值±SD.number of exacerbations was 0.6±0.9. Equivalent data for the treatment arms were 0.6±0.8 for ciprofloxacin DPI 14 days on/off compared with 0.7±1.0 in the matching placebo arm and 0.4±0.6 for ciprofloxacin DPI 28 days on/off与0.7±1.1 in the matching placebo arm.
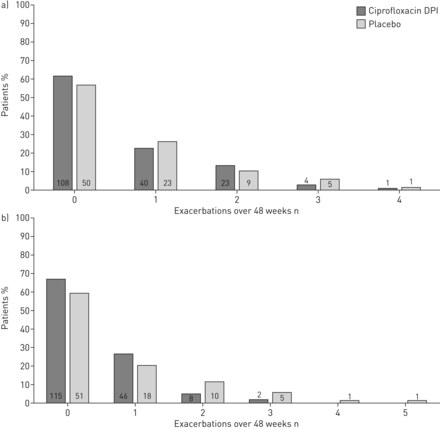
对于吸入的环丙沙星干粉(DPI)A)14天接通或匹配安慰剂和B)28天,接通或匹配安慰剂或匹配安慰剂或匹配安慰剂或匹配安慰剂或匹配安慰剂,或匹配安慰剂或匹配安慰剂的患者,为48周的频率。在酒吧内或上方的数字表示患者的数量。对于主要的终点,需要达到三个标准:1)在至少三个症状或症状(呼吸困难,喘息,咳嗽,24-h痰体积或痰脓)超出正常日常变异的标准中至少2天,2)发烧(体温> 38.0°C)或萎靡不振,3)全身抗生素治疗。
Over 48 weeks, 61.4% of patients treated with ciprofloxacin DPI 14 days on/off compared with 67.3% of patients treated with ciprofloxacin DPI 28 days on/off and 58.0% of patients in the pooled placebo arm did not experience a protocol-defined exacerbation for the primary end-point.
Subgroup analyses
Exploratory analyses of the primary end-point in pre-specified subgroups did not reveal any differences in any of the subgroups for time to first exacerbation or frequency of exacerbations (补充图S1)。
次要终点
由于统计分析计划的分层性质,因此不能对次要终点进行正式的重要性测试,因为未达到主要终点。二次终点的探索性分析表明,环丙沙星DPI治疗与益处有关,包括降低的加剧频率(根据主要终点定义)与pooled placebo for the ciprofloxacin DPI 28-day on/off arm (figure 4)。

结果对美国食品和药物管理局(FDA)和欧洲药品局(EMA)/其他机构的欧洲药物局(EMA)/其他机构对CIPROFloxacin DPI A)14天,B)28天开/关与汇集安慰剂(PPL) or matching placebo (MPL). HR: hazard ratio; IRR: incidence rate ratio; EOT: end of treatment; SGRQ: St George's Respiratory Questionnaire; QOL-B RSS: Quality of Life-Bronchiectasis respiratory symptoms domain score; FEV1: forced expiratory volume in 1 s.#: the primary (stringent) definition of exacerbation was a respiratory event that met three separate criteria: 1) worsening in at least three signs or symptoms (dyspnoea, wheezing, cough, 24-h sputum volume or sputum purulence) beyond normal day-to-day variation for at least 2 consecutive days, 2) fever (body temperature >38.0°C) or malaise/fatigue, and 3) systemic antibiotic treatment;¶: the less stringent definition of exacerbation was a respiratory event with worsening of at least one of the aforementioned signs or symptoms and systemic antibiotic use;+:Pseudomonas aeruginosa,嗜血杆菌流感,Moraxella catarrhalis,Staphylococcus aureus,Streptococcus pneumoniae,Stenotrophomonas maltophiliaorBurkholderia cepacia(eradication of pathogens was defined as a negative culture result at EOT for all pre-specified pathogens in a subject with a positive baseline culture for at least one pre-specified pathogen; occurrence of a new pathogen was defined as a positive culture for at least one pre-specified pathogen at EOT in a subject who did not have a positive culture for that pathogen at baseline). Dashed lines indicate statistical significance was not reached. The α level for end-points was 0.049 for the 14-day arm and 0.001 for the 28-day arm. The confidence interval is 95.1% for comparisons of ciprofloxacin DPI 14 days on/off与placebo and 99.9% for comparisons of ciprofloxacin DPI 28 days on/off与placebo.
The mean±SD.number of exacerbations reported using the less stringent definition was 0.7±1.0 for ciprofloxacin DPI 14 days on/off compared with 0.8±1.1 in the matching placebo arm and 0.5±0.8 for ciprofloxacin DPI 28 days on/off compared with 0.9±1.1 in the matching placebo arm.
Safety
环丙沙星DPI耐受良好,在CiProfloxacin DPI 28天ON / OFF臂中观察到最低的不良事件速率。519名(67.2%)患者共有349名(67.2%)在注册后的任何时间经历了不良事件:131(75.3%)患者在Ciprofloxacin DPI 14天开/关臂,101(59.1%)中的Ciprofloxacin DPI 28- 在汇集安慰剂集团中on / OFF ARM和117(67.2%)。
总体而言,64.9%的患者经历了治疗紧急的不良事件。治疗紧急肌肉骨骼不良事件令人震惊。两名患者在每次Ciprofloxacin DPI治疗组(一个肌腱紊乱和一个触发手指在Ciprofloxacin DPI 14天/关闭手臂中的一个患者报告肌腱疾病;在Ciprofloxacin DPI 28天开/关臂中的不适和一个韧带扭伤)安慰剂组中的零患者。由于治疗 - 紧急不良事件引起的过早的停止罕见,并在治疗臂的类似频率发生(table 4)。
Treatment-emergent adverse events (TE-AEs)#(MedDRA classifications)
Serious treatment-emergent adverse events occurred in 22.0% of patients overall; rates were slightly lower in the ciprofloxacin DPI 28-day on/off arm compared with other arms. Respiratory disorders and infections/infestations were the most common system organ classes reported for serious treatment-emergent adverse events (补充表S4)。One serious treatment-emergent adverse event (atrial flutter) was considered drug related. This treatment-emergent adverse event occurred in a subject in the ciprofloxacin DPI 28-day on/off arm who had a history of ischaemic heart disease. This patient also experienced severe fatal congestive cardiomyopathy, which was reported in parallel but was not considered to be drug related.
Nine treatment-emergent deaths were reported during the study (three (1.7%) in the ciprofloxacin DPI 14-day on/off arm, four (2.3%) in the ciprofloxacin DPI 28-day on/off arm and two (1.1%) in the pooled placebo group), but no fatalities were considered to be drug related and deaths were consistent with morbidities common to the patient population being studied (补充表S4)。
张es in ciprofloxacin minimal inhibitory concentrations
At baseline, 98 out of 521 (18.8%) patients had a pathogen with an elevated minimal inhibitory concentration (MIC) for ciprofloxacin (defined as resistant based on systemic ciprofloxacin breakpoints as specified in补充部分S2) isolated from their sputum. The number of patients with the development of at least one isolate from sputum with an elevated MIC from pre-treatment to any time point during the study was 37 out of 176 (21.0%) for ciprofloxacin DPI 14 days on/off, 28 out of 170 (16.5%) for ciprofloxacin DPI 28 days on/off and 17 out of 173 (9.8%) for pooled placebo. At the end-of-study (EOS) visit that occurred 8 weeks after the end of treatment, pathogens with elevated MICs were isolated from 26 patients (5%) (ciprofloxacin DPI 14 days on/off 12 out of 176 (6.8%), ciprofloxacin DPI 28 days on/off 10 out of 171 (5.8%) and pooled placebo four out of 174 (2.3%)) who had susceptible bacteria at baseline. During the study, elevated MICs were more common in patients receiving active therapy. The percentage of patients with at least one isolate from sputum with an elevated MIC at any time point including baseline and EOS was 52.3% for ciprofloxacin DPI 14 days on/off, 39.8% for ciprofloxacin DPI 28 days on/off and 29.3% for pooled placebo.
讨论
The RESPIRE 2 trial constitutes the second trial in the RESPIRE programme. RESPIRE 1 demonstrated a significant treatment effect for the ciprofloxacin DPI 14-day on/off treatment regimen and a trend towards improved outcomes in the ciprofloxacin DPI 28-day on/off arm [8]。In RESPIRE 2, end-points favoured ciprofloxacin DPI over placebo, but the positive efficacy signals did not translate into statistical significance for either active treatment arm. The observed positive trends in RESPIRE 2 were generally more pronounced in the ciprofloxacin DPI 28-day on/off arm than in the 14-day on/off arm. However, due to the pre-specified significance level of 0.001 for the ciprofloxacin DPI 28-day on/off arm, differences from placebo did not achieve statistical significance.
呼吸2的加热速率远低于预期,低于呼吸中观察到的那些1.在呼吸2中,62.2%的患者没有根据主要定义(由三个症状或症状定义)的恶化,这是显着的比呼吸1患者的52%没有加剧[8]。As the study entry criteria were chosen to enrich the population of “frequent exacerbators”, requiring two or more documented exacerbations in the previous year, the low exacerbation rates that occurred during the trial were unexpected. For both trials, a stringent definition of exacerbation was used for primary end-point evaluations [9]。This definition was not, however, applied for the entry criteria. As each patient experiences their disease differently [10] and as patients are often educated to take antibiotic treatment when symptoms increase, it is likely that the prior exacerbations did not meet the stringent definition used during the trial. A proposed standardised definition of an exacerbation has recently been published [11]; this may be of benefit in future clinical trials to reduce heterogeneity.
Understanding the low exacerbation rates observed in both active and placebo arms of RESPIRE 2, and the differences in exacerbation rates between RESPIRE 1 and 2, requires examination of a number of factors, including patient characteristics and geography. Baseline demographics varied somewhat between RESPIRE 1 and RESPIRE 2, despite their identical inclusion and exclusion criteria. As patient phenotype is known to affect prognosis [10],不同的患者型材可能导致不同的临床结果。呼吸患者2普遍较小(平均年龄60.1与64.7 years) and more likely to have had only two exacerbations in the last year (78%与55%)比呼吸患者1 [8]; two episodes was the minimum frequency of exacerbations required to enter the study. RESPIRE 2 patients had poorer lung function as indicated by the proportion of patients with FEV1% pred <50% (41.8%与由St George的呼吸问卷调查症状成分分数评估30.0%)和更多的呼吸系统症状(平均61.0与56.8) [8]。
Although a primary diagnosis of COPD was an exclusion criterion, 28% of patients in RESPIRE 2 had a history of COPD与呼吸1中16%,表明呼吸2中的更多患者对支气管扩张/ COPD的重叠诊断。这是呼吸2(8.1%)的较大的茶碱使用与呼吸1(2.2%)。COPD和支气管扩张之间关系的确切性质仍有待建立。这些患者的最佳治疗很难理解,可能与单独任何病症推荐的疗法不同[12]。排除患有重叠支气管扩张/ COPD诊断的患者可能导致临床试验中更均匀的NCFB患者群体。呼吸患者2具有更严重的气流阻塞的指标,但无法充分评估支气管扩张严重程度,因为计算断层扫描扫描未在基线上进行评估。并非所有包括在Bronchiectasis严重性指数中的所有参数都被收集在基线上,因为这些细节尚未在设计呼吸研究时出版[7,13]。
RESPIRE 1 and 2 also differed in the geographical regions that were represented, with RESPIRE 2 enrolling more patients from Asia and Eastern Europe. The variability in clinical practice could have potentially impacted the determination of exacerbations and treatment prior to enrolment. Specialist care for individuals with bronchiectasis is well developed, and research interest is relatively intense in Western Europe, the USA and Australia, which has led to the development of treatment guidelines and national standards [14–16]。European guidelines have only recently been published [17]类似的举措在世界其他地区的情况下不太普遍[1]。这可能导致入学前的不同疾病管理实践。例如,呼吸2中的患者不太可能用标准呼吸药物治疗(67.9%)与80.5%) or long-term macrolide treatment (7.7%与15.9%) than those in RESPIRE 1 [8]。这可能反映了个别国家实践,或者可以通过不同的患者表型解释。在入学前的表型差异和护理标准方面减少患者人群的异质性,是未来试验的重要考虑因素。
Any comparison of the outcomes of RESPIRE 1 and 2 should take into consideration the change in extrapolation method and significance levels. Based on experience gained in RESPIRE 1, significance levels were amended by the sponsor with agreement from regulatory authorities to maximise the likelihood of achieving a significant result in the ciprofloxacin DPI 14-day on/off arm. If the α adjustment applied in RESPIRE 1 (p=0.025) had been used in RESPIRE 2, the ciprofloxacin DPI 28-day on/off arm would have met the primary end-point for the EMA/others analysis of frequency of exacerbations (IRR 0.55, 99.9% CI 0.30–1.02; p=0.0014). The FDA primary end-point, however, would not have been statistically significant nor would results in the ciprofloxacin DPI 14-day on/off arm have been affected.
如在呼吸1中所观察到的,环氟氯酰嘧啶DPI良好耐受。相关的研究药物相关的治疗急性不良事件在呼吸2中显着降低,而不是呼吸1(13.7%)与25.1%)[8]。Differences were largely due to lower numbers of respiratory treatment-emergent adverse events; contributory factors may include a lower rate of exacerbations in RESPIRE 2 or a higher threshold for diagnosis of these adverse events in centres involved in RESPIRE 2与RESPIRE 1. Trial discontinuation rates were low (5% due to treatment-emergent adverse events) and comparable across treatment arms. Together with data from RESPIRE 1, these findings support a favourable safety profile for ciprofloxacin DPI.
The limitations of the RESPIRE programme study design have been discussed previously [8]。RESPIRE 2 additionally highlighted the impact of low exacerbation rates on efficacy outcomes. Future NCFB trials should consider using a more stringent definition for prior exacerbations as an inclusion criterion and collecting data needed to calculate bronchiectasis severity based on published severity assessment tools [7,13]。
总之,尽管效果显示tr信号ends towards clinical benefits and excellent tolerability in the RESPIRE 2 study, neither the 14- or 28-day on/off ciprofloxacin DPI arms met the primary end-points of increasing time to first exacerbation or reducing frequency of exacerbations. The RESPIRE clinical trial programme thus encapsulates both the promise and difficulties of documenting a statistically significant impact on exacerbations in NCFB patients, and substantially contributes to the body of knowledge on the optimal design of clinical trials.
Supplementary material
披露
补充材料
T. AksamitERJ-02053-2017_Aksamit
T-J。班塞尔ERJ-02053-2017_Bandel
A. De SoyzaERJ-02053-2017_De_Soyza
J.S. ElbornERJ-02053-2017_Elborn
E. Operschall.ERJ-02053-2017_OPERSCHALL.
E. Polverinoerj - 02053 - 2017 - _polverino
K. RothERJ-02053-2017_Roth
R. WilsonERJ-02053-2017_Wilson
K.L.Winthrop.ERJ-02053-2017_WINTHROP.
致谢
We thank the patients, investigators (补充部分S4) and study centres that contributed to RESPIRE 2. We also thank Ulrike Krahn (Bayer AG, Wuppertal, Germany) and Elaine Montegriffo (Bayer PLC, Reading, UK) for assisting with statistical analysis; Jeff Alder (Bayer US LLC, Whippany, NJ, USA) for microbiology data assessment and critically reviewing the data; Maxine Lau (Bayer AG, Berlin, Germany) for pharmacovigilance input; and Fusion MD (Montreal, Canada) and highfield:communication (Oxford, UK) for providing medical writing services with funding from Bayer AG. A. De Soyza, R. Wilson and J.S. Elborn acknowledge the support of EMBARC and the Medical Research Council-funded BRONCH-UK collaborative for peer support and advice in bronchiectasis.
Footnotes
This article has supplementary material available fromwww.qdcxjkg.com
This study is registered atClinicalTrials.gov使用标识符NCT02106832。
Support statement: This study was supported by Bayer AG. Funding information for this article has been deposited with theCrossRef Resder注册表。
Conflict of interest: Disclosures can be found alongside this article atwww.qdcxjkg.com
- ReceivedOctober 6, 2017.
- Accepted2017年11月20日。
- Copyright ©ERS 2018











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}