





抽象的
继发性细菌性肺炎常加重甲型流感病毒(IAV)感染的严重性和致死性。然而,引起死亡率增加的人类肺组织机制尚不清楚,缺乏治疗性免疫调节选择。
我们建立了一个人的肺离体采用共感染模型,研究导致继发性肺炎球菌肺炎易感性的先天免疫相关机制。
我们发现I型和III型干扰素(IFN)有抑制作用肺炎链球菌肺炎料诱导白细胞介素(IL)-1β释放。IL-1β的缺乏导致了细菌诱导的粒细胞-巨噬细胞集落刺激因子(GM-CSF)释放的抑制。特异性抑制IFN受体I和iii相关酪氨酸激酶2 (Tyk2)完全恢复S.肺炎-诱导IL-1β-GM-CSF轴,导致细菌生长减少。先前的IAV感染人类肺泡导致I型和III型ifn依赖的早期细胞因子IL-1β和GM-CSF的阻断,这是协调针对细菌的充分先天免疫反应的关键。病毒诱导的抑制可能导致细菌清除和肺泡修复受损。
TYK2的药理抑制可能是在IAV相关的二次细菌肺炎中维持有益内源性GM-CSF水平的新治疗选择。
抽象的
Tyk2抑制可能是维持严重病毒/细菌肺炎中内源性GM-CSF水平的策略http://ow.ly/Owv130aWr6Z
介绍
严重肺炎在世界范围内造成很高的死亡率,自引入抗生素以来死亡率几乎没有变化[1,2].甲型流感病毒(IAV)和肺炎链球菌肺炎料,特别是在随后的合并感染中,占大多数致命结果[3.].考虑到继发性细菌性肺炎造成的死亡人数、未来禽流感暴发的威胁以及细菌对抗生素日益增强的耐药性[4]因此,了解病毒、细菌和肺靶细胞之间的分子相互作用至关重要,从而使创新的辅助治疗超越病原体导向的临床方法。
先前的小鼠研究表明,继发性细菌联合感染加重的机制包括iav诱导的吞噬细胞功能改变、增加细菌粘附或改变免疫系统成分的上皮损伤[4].特别是,iav诱导的I型和II型干扰素(IFNs)被怀疑调节先天免疫反应,如趋化因子(C-X-C基序)配体[5]趋化因子、白细胞介素(IL)-17产生γδT细胞或继发性细菌感染的细菌清除[6,7]然而,IAV诱导的IFN对人类肺组织中次级细菌病原体识别的干扰是完全未知的。
在体外关于单核细胞和巨噬细胞的研究表明,IFN可能干扰中央炎症细胞因子途径,例如炎症,从而抑制IL-1β的生产[8- - - - - -10.].反过来,IL-1β似乎对生存至关重要S.肺炎感染或感染后IAV金黄色葡萄球菌小鼠的肺炎[11.,12.].细胞源,分子机制及其在人肺中诱导后续细胞因子反应的作用仍然未知。
除了IL-1β,IFN改变了生产用于肺部炎症控制另一个中央因子,粒细胞 - 巨噬细胞集落刺激因子(GM-CSF)[13.,14.].在感染IAV(H1N1)的小鼠中,GM-CSF增强了肺泡巨噬细胞的量和抗性,以及树突细胞的募集和激活,两者都显着促进生存率[15.- - - - - -18.].同样,在小鼠中,由于肺泡巨噬细胞增强了对细菌的杀灭作用,死亡率得到了改善S.肺炎或B组链球菌,甚至用革兰氏阴性感染肺炎克雷伯菌GM-CSF处理后表现出较少的细胞凋亡和肺泡渗漏[19.- - - - - -21.].此外,首个使用GM-CSF辅助治疗急性肺损伤或成人呼吸窘迫综合征(ARDS)的临床试验强调了这种分子的治疗潜力[22.,23.].
因此,我们建立了离体IAV和IAV的人肺共感染模型S.肺炎研究这些细胞因子在人肺泡腔内的细胞调节作用。我们的研究在细胞水平上证明了iav诱导的IFN是如何阻碍的S.肺炎[相关的IL-1β释放,从而抑制人肺泡中的GM-CSF生产。此外,通过抑制IFN受体相关的酪氨酸激酶2(Tyk2),我们提供潜在的药理学治疗选择,以恢复对二次细菌感染的病毒抑制免疫应答。
材料和方法
方法
在线补充材料中概述了详细信息。
人肺组织
正常的外周人肺组织是从患有支气管癌的患者身上新获得的。该研究得到了柏林Charité–Universitätsmedizin大学伦理委员会的批准(项目EA2/050/08和EA2/023/07);获得了所有患者的书面知情同意。该组织被冲压成小圆柱体(3×8×8) mm)并按照所述进行培养[24.].
原代人肺泡巨噬细胞和肺泡上皮细胞的分离
用Hanks平衡盐溶液(HBSS)反复灌注人肺组织,分离肺泡巨噬细胞,并将其接种在6个孔板(1×10)中6细胞)并在RPMI 1640培养基中培养2-4天 天。II型肺泡上皮细胞(AECs)采用Elet al。[25.].简单地说,去除肺泡巨噬细胞后,将人肺组织剁碎,用胰蛋白酶I型和脱氧核糖核酸酶消化。消化后的组织通过无菌纱布过滤器连续过滤。细胞在1:1 HBSS:杂杂瘤-无血清培养基中离心并孵育1.5小时,以区分肺泡巨噬细胞的粘附。随后,收集细胞碎片,离心并在Pancoll (PanBiotech, Aidenbach, Germany)不连续梯度上分层。离心后,收集界面层细胞,用HBSS洗涤,播种于24孔板,RPMI 1640培养。
病毒和细菌株
如W的描述,人季节性IAV菌株A / PANAMA / 2007/1999(PAN / 99(H3N2))传播艾因海默et al。[26.].两个菌株的S.肺炎如前所述,使用和培养封装菌株D39血清型2(NCTC7466)和临床分离血清型3(ST3,SN35209)[27.].
人肺组织和细胞感染和刺激
在感染实验中,首先将肺培养接种IAV (1×106PFU)处理24 h,再用S.肺炎(D39或ST3,1×106CFU·毫升−1)如图所示。含有重组IFN-β、-γ、-λ的培养基1, IFN-β和-γ的组合或IFN-β、-γ和-λ的组合1(100 U·mL−1肿瘤坏死因子(TNF)-α (100 U·mL)−1)、IL-1β (10 ng·mL .−11µg·mL−1)或PRT2070(cerdulatinib)(1 在指定的时间点将µM)注射到肺外植体中。对照外植体接受等量的含有PBS的培养基。AEC II和肺泡巨噬细胞感染S.肺炎D39(1多种感染(MOI))16小时。此外,AEC II用IL-1β刺激(5 ng·mL−1) 16小时。在肺泡巨噬细胞上清实验中,用阿纳金菌(1µg·mL)预处理AEC II−1)和肺泡巨噬细胞上清液(16 hS.肺炎加入D39,10II)并孵育20小时。
结果
人肺组织模型离体IAV和IAV的共同感染S.肺炎
我们建立了一个混合感染模型离体用对照培养基或Pan/99(H3N2)接种培养的人肺组织24小时 h(在线补充图S1A和S1B)。随后,使用S.肺炎D39 for a further 16 h(在线补充图S1C和S1D)。如前所述,由前表面活性剂蛋白C共定位显示,只有AEC II被IAV感染[26.].S.肺炎检测到D39紧密附着在AEC I和AEC II的细胞表面以及肺泡巨噬细胞上,不依赖于先前的IAV感染(在线补充图S1D)。
iav诱导的I、II和III型IFN在合并感染时表达未发生变化S.肺炎
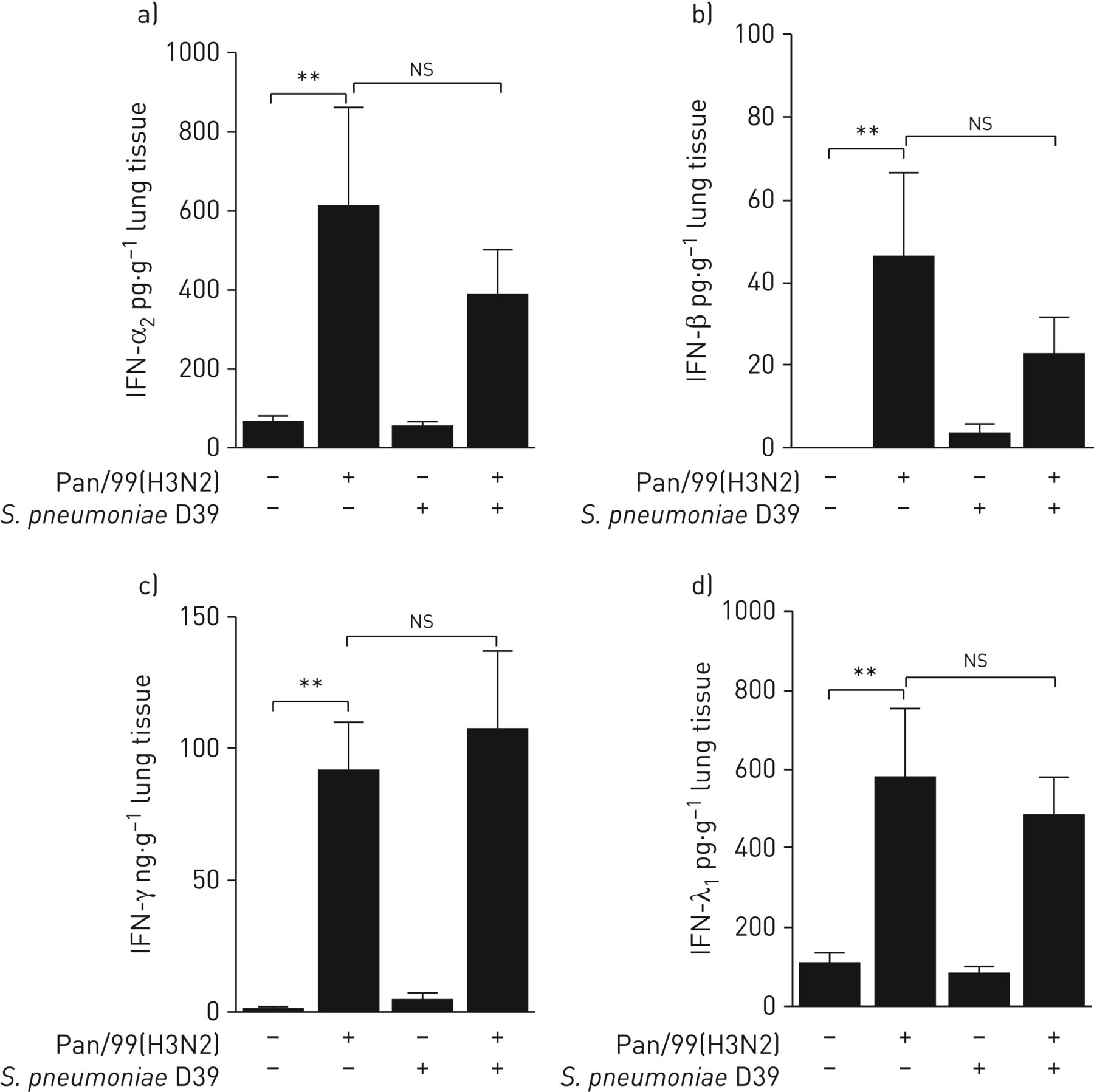
假设IFNS在对二次细菌感染易感性中发挥重要作用;然而,这仍未对人肺组织未探明。因此,我们首先分析了在肺组织上清液中单一和共感染后不同IFN类型的分泌。PAN / 99(H3N2)显着诱导IFN-α的释放2IFN-β(Ⅰ型)、IFN-γ(Ⅱ型)和IFN-λ1(III类),鉴于S.肺炎D39完全没有IFN诱导(图1一个- d)。所有IFN的分泌模式在病毒和细菌联合感染中没有明显改变,说明S.肺炎对IFN的调节没有影响,这可能为受损的第二宿主防御细菌铺平道路。
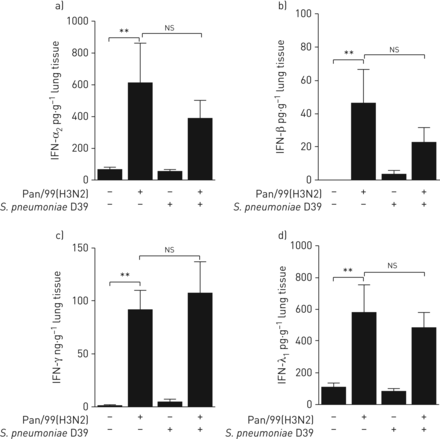
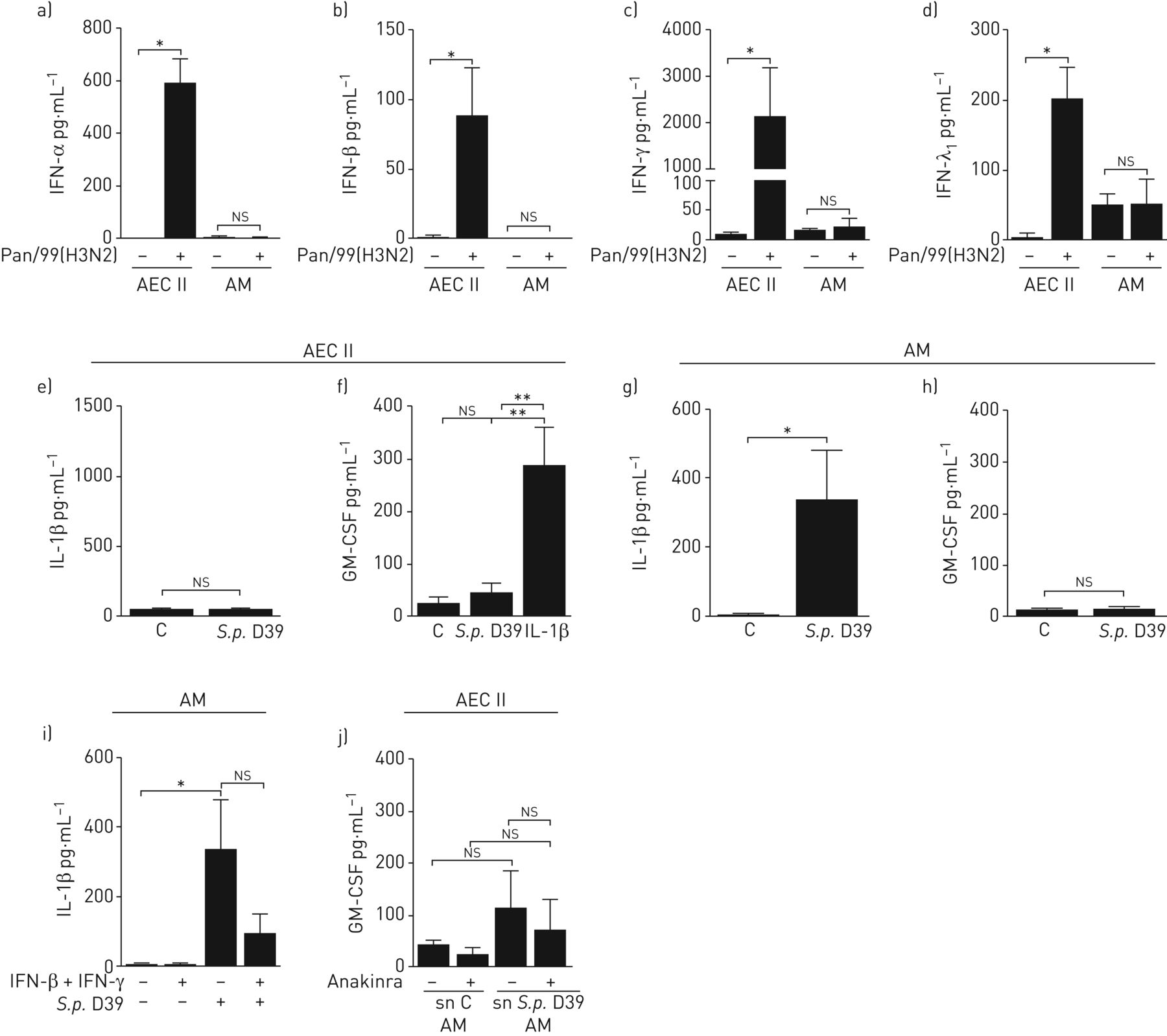
干扰素(IFN)共同感染的人肺组织生产。肺外植体离体模拟感染或挑战甲型流感病毒Pan/99(H3N2)(1×106 PFU·mL−1).24 h后,将专用外植体侵染肺炎链球菌肺炎料D39(1×106CFU·毫升−1)或模仿感染。肺炎球菌感染16小时后收集上清,检测a) I型IFN-α2;b)干扰素-β;c) II型IFN-γ;和d) III型IFN-λ1发布。数据表示为平均值±扫描电镜在六个独立实验的捐献者中ns:不重要。**:p≤0.01.
IAV和IFN会干扰S.肺炎- 诱导人肺中的IL-1β-GM-CSF轴
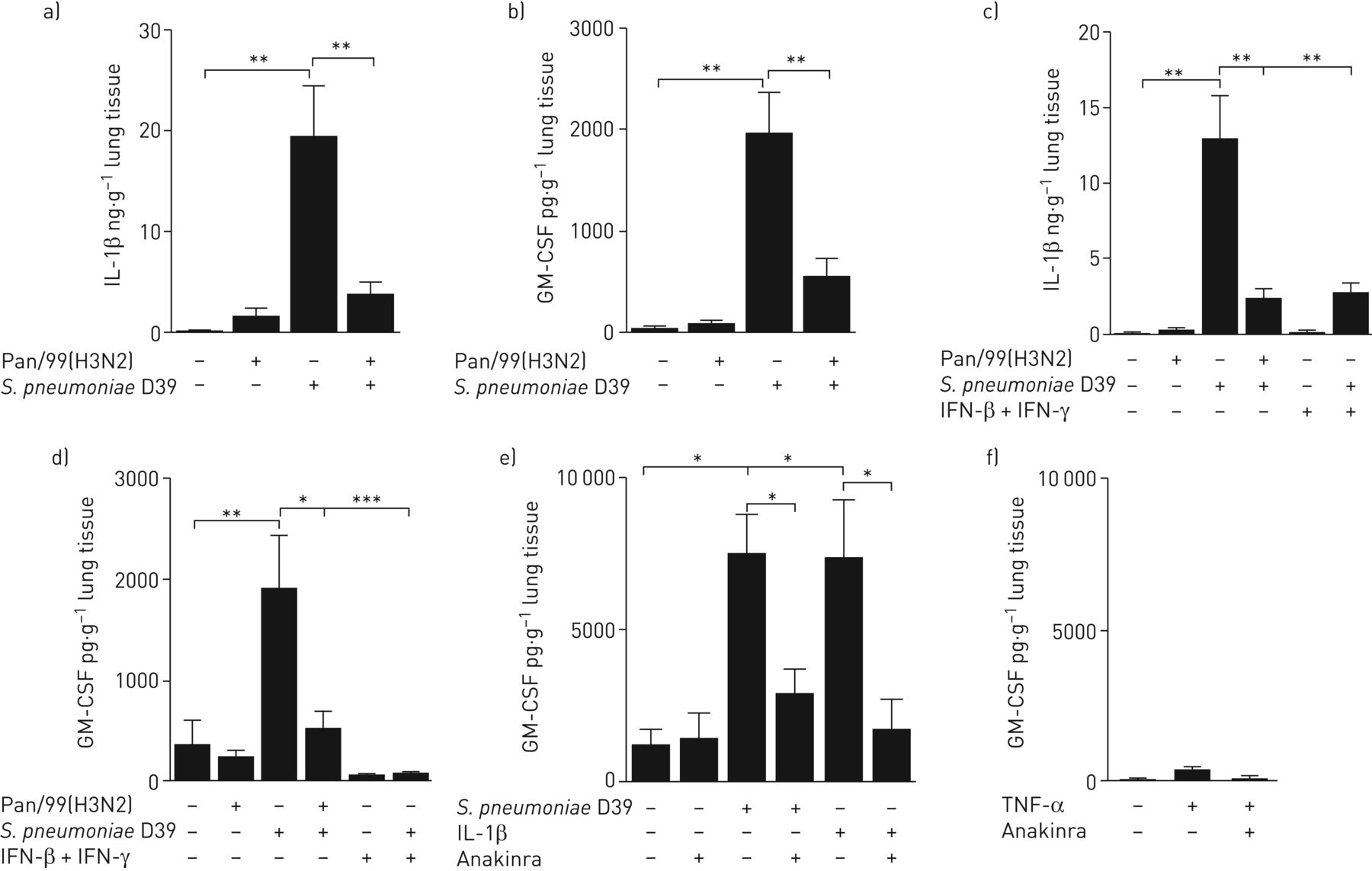
细胞因子和趋化因子的诱导在原先生免疫反应中起到对照病毒和细菌感染的起始作用重要作用[28.]因此,对小鼠的研究表明,在原发性非致死性流感病毒感染期间诱导I型和II型IFN使对一系列细菌病原体的防御变得复杂[6,7].然而,它们在人类肺部合并感染方面的作用迄今尚不清楚。我们首先研究了IAV对人肺上清液中典型肺炎球菌诱导因子调控的影响。S.肺炎D39感染显着诱导所有测试的细胞因子,但在IAV存在下,在IAV的情况下在IAV-6,IL-8,IL-10和TNF-α的共感染中没有发现变化(在线补充图S2A-D)。但是,前IAV感染显着降低S.肺炎D39诱导IL-1β和GM-CSF(图2ab),证实为临床ST3分离株(在线补充图S2E和F)在体外研究表明,IFN可能会干扰IL-1β和GM-CSF诱导,我们测试了人肺组织中的IFN是否可以模仿IAV诱导的IL-1β和GM-CSF [8- - - - - -10.,13.,14.].其次是Pan/99(H3N2)单次感染和合并感染S.肺炎D39,我们用IFN-β和IFN-γ进行的IAV取代了同一时间课程,并比较了IL-1β和GM-CSF的释放量。根据IAV感染,IFN显着阻止S.肺炎诱导IL-1β和GM-CSF合成(图2 c有趣的是,Cakarovaet al。[29.表明小鼠肺泡上皮GM-CSF的表达依赖于TNF-α的诱导。我们观察到与IL-1β (图2ac),在IAV感染或用IFN的治疗期间,肺炎球菌诱导的TNF-α的水平保持不变(在线补充图S3)。因此,我们假设在人肺组织中,GM-CSF的表达依赖于IL-1β而不是TNF-α。在16-H之前用IL-1受体拮抗剂Anakinra(4h)预处理人肺S.肺炎D39感染或IL-1β刺激显著降低GM-CSF表达(图2 e)相反,TNF-α刺激人肺不足以诱导GM-CSF(图2 f).在同一肺中诱导环氧化酶-2作为有效TNF-α刺激的阳性对照(在线补充图S4A)。
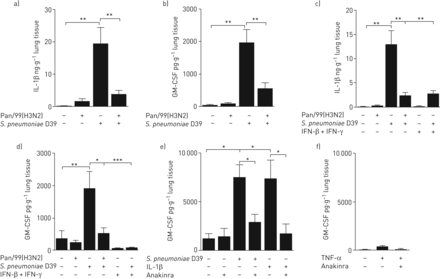
甲型流感病毒Pan/99(H3N2) (IAV)联合感染和干扰素(IFN)治疗可抑制肺炎链球菌肺炎料D39诱导人肺中的白细胞介素(IL)-1β-粒细胞 - 巨噬细胞菌落刺激因子(GM-CSF)轴。肺外植体离体模拟感染或挑战(a和b) IAV (1×106 PFU·mL−1或(c-e) IFN-β和IFN-γ联合(100 U·mL−1每人)16人 H然后,将专用样本感染病毒S.肺炎(1×106CFU·毫升−1).肺炎球菌感染16小时后收集上清,检测a) IL-1β的释放;b) gm - csf;c) il - 1β;d) gm - csf。e)用IL-1β受体拮抗剂anakinra (1 ng·mL)处理肺组织−1)或对照培养基。4天后 h、 显示的肺标本要么模拟感染,要么用S.肺炎(1×106CFU·毫升−1)或用重组IL-1β刺激(10ng·ml−1),然后测定GM-CSF释放量。f) Lung tissue was either mock stimulated or treated with tumour necrosis factor (TNF)-α (100 ng·mL−1),在有无阿纳金菌(1ng·mL .)作用16 h−1).4 h后,对肺标本进行模拟感染或激发S.肺炎(1×106CFU·毫升−1),然后测定GM-CSF释放量。数据以平均值±表示扫描电镜至少有四名捐赠者参与了独立实验。*: p≤0.05;**:P≤0.01。
为了强调GM-CSF释放对IL-1β产生的直接依赖性,我们测定了这两个因素的时间进程S.肺炎-感染的人肺组织。表达模式的直接比较显示,在4-6小时后,从大量IL-1β开始的时移分泌 8-16小时后,接着是GM-CSF h(在线补充图S4B)。综上所述,这些数据表明,人肺中GM-CSF的释放主要依赖于IL-1β而不是TNF-α。
AEC II和肺泡巨噬细胞来源的IFN阻断肺泡巨噬细胞的IL-1β,从而抑制AEC II表达的GM-CSF
我们的数据表明,IAV诱导的IFN阻断了IL-1β的释放,最终导致人肺中GM-CSF生成的丧失。为了进一步研究这种细胞因子调节在病毒和细菌共同感染中的潜在细胞相互作用,我们从新鲜人肺组织中分离肺泡巨噬细胞和AEC II。免疫荧光染色的表型特征显示分离的肺泡巨噬细胞对CD68、AEC II对泛细胞角蛋白和前表面活性剂蛋白C呈阳性(在线补充图S5A和S5B)。首先,我们研究了IAV感染的AEC II和肺泡巨噬细胞释放IFN的情况,证明这两种细胞类型都产生IFN-α、-β和-λ1,但IFN-γ主要由AEC II在人肺泡腔内释放(图3a-d).相反,AEC II在感染后IL-1β的释放呈阴性S.肺炎D39(图3 e),但在IL-1β处理后显示出GM-CSF的显着释放。符合完整的人肺组织的刺激,S.肺炎在AEC II中未能直接诱导GM-CSF分泌(图3 f).相比之下,S.肺炎肺泡巨噬细胞的感染导致IL-1β的强烈释放,但未诱导GM-CSF表达(图3 g和h)。同时用I型和II型干扰素治疗肺泡巨噬细胞足以抑制IL-1β的产生(图3我),而TNF-α在相同的样本中不降低(在线补充图S6),镜像在完整的肺组织中获得的结果。
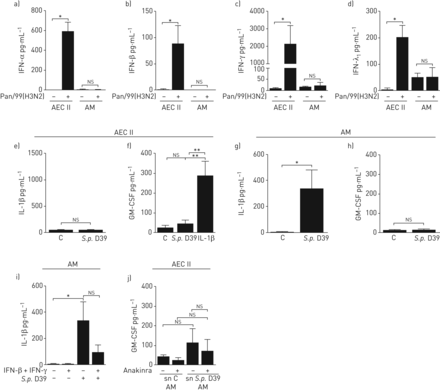
甲型流感病毒Pan/99(H3N2) (IAV)感染后,肺泡上皮II型细胞(AEC II)和肺泡巨噬细胞(AM)产生I、II和III型干扰素(IFN),抑制AM诱导的白细胞介素(IL)-1β,从而抑制AEC产生的粒细胞-巨噬细胞集落刺激因子(GM-CSF)。AEC II和AM均从新鲜人肺组织中分离,分别培养3-4天(AEC II)和2天(AM)。为了识别IFN的细胞来源,a) IFN-α的释放;b)干扰素-β;c)干扰素-γ;和d)干扰素-λ1(1×106 PFU·mL−1; 24 h) 感染细胞与模拟感染细胞的比较。e)在AEC II中检测IL-1β和f)GM-CSF,用肺炎链球菌肺炎料(标准普尔。)D39(1多种感染(MOI))16小时。f)与IL-1β的AEC II的额外刺激(5 ng·ml−1)显示GM-CSF诱导AEC II。g)检测IL-1β和h) GM-CSFS.肺炎(教学语言1)16人 Hi) 用IFN-β和IFN-γ(100)联合模拟激发或刺激AM U·mL−1each) 16 h before pneumococcal infection and supernatants were assayed for release of IL-1β. j) AEC II were treated with anakinra (1 ng·mL−1)或控制介质,用于4 H然后将细胞培养20分钟 用对照(C)培养基激发AM获得的上清液(sn)或S.肺炎(教学语言1分16秒) h) .对AEC II的上清液进行GM-CSF释放分析。数据以平均值±表示扫描电镜至少有三名捐赠者参与了独立实验。ns:不重要。*:p≤0.05;**:p≤0.01.
最后,肺泡巨噬细胞上清液感染S.肺炎用于在有或无阿纳金拉的AEC II中刺激GM-CSF,为上皮GM-CSF依赖肺泡巨噬细胞释放的IL-1β提供证据(图3 j).然而,上皮GM-CSF表达的抑制似乎并没有被限制为仅在肺泡巨噬细胞IL-1β抑制;代替IFN也可以直接抑制IL-1β刺激(在线补充图S7)后GM-CSF。
TYK2抑制还原IAV诱导的I型和III IFN介导的IL-1β-GM-CSF轴的抑制,并减少细菌生长
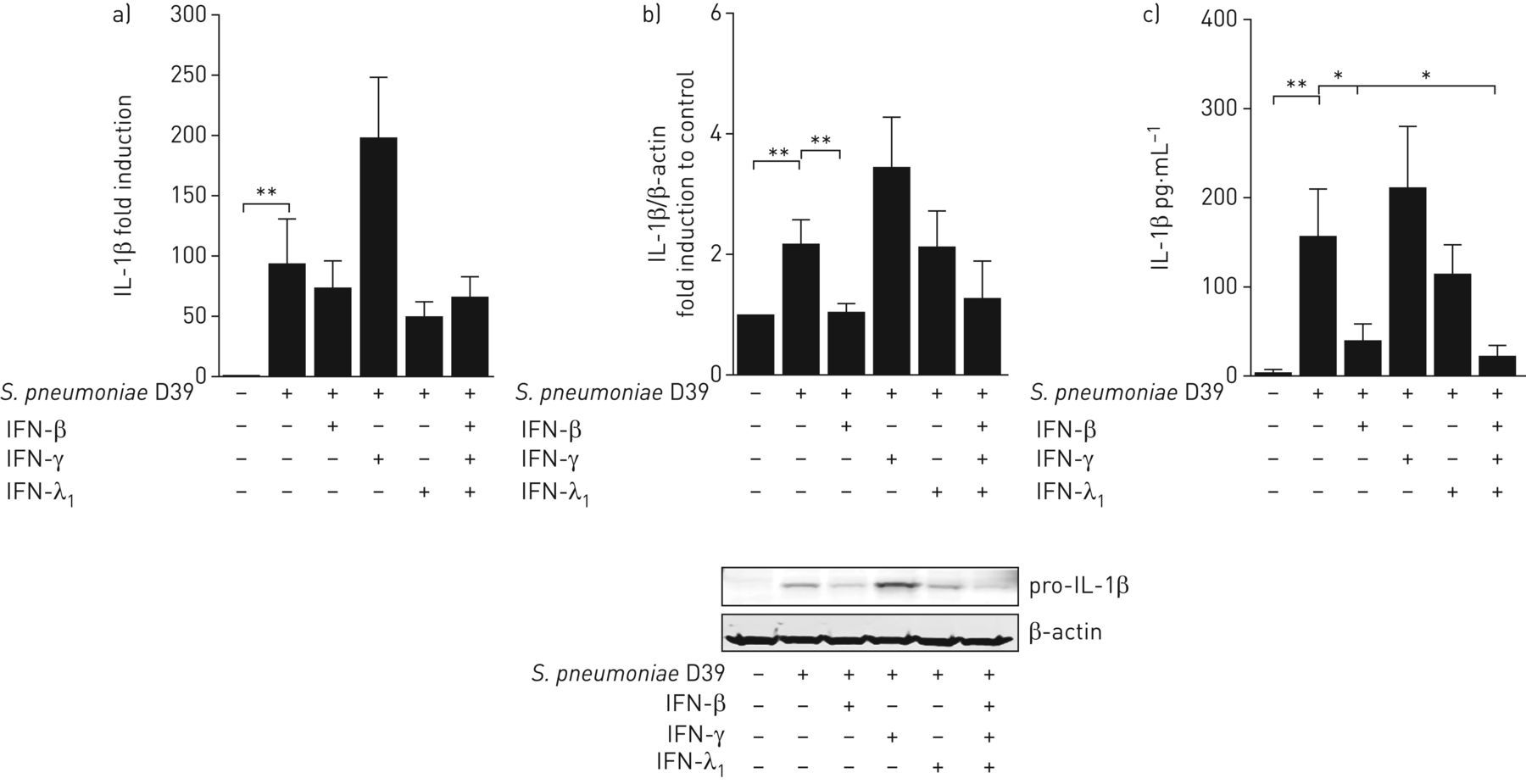
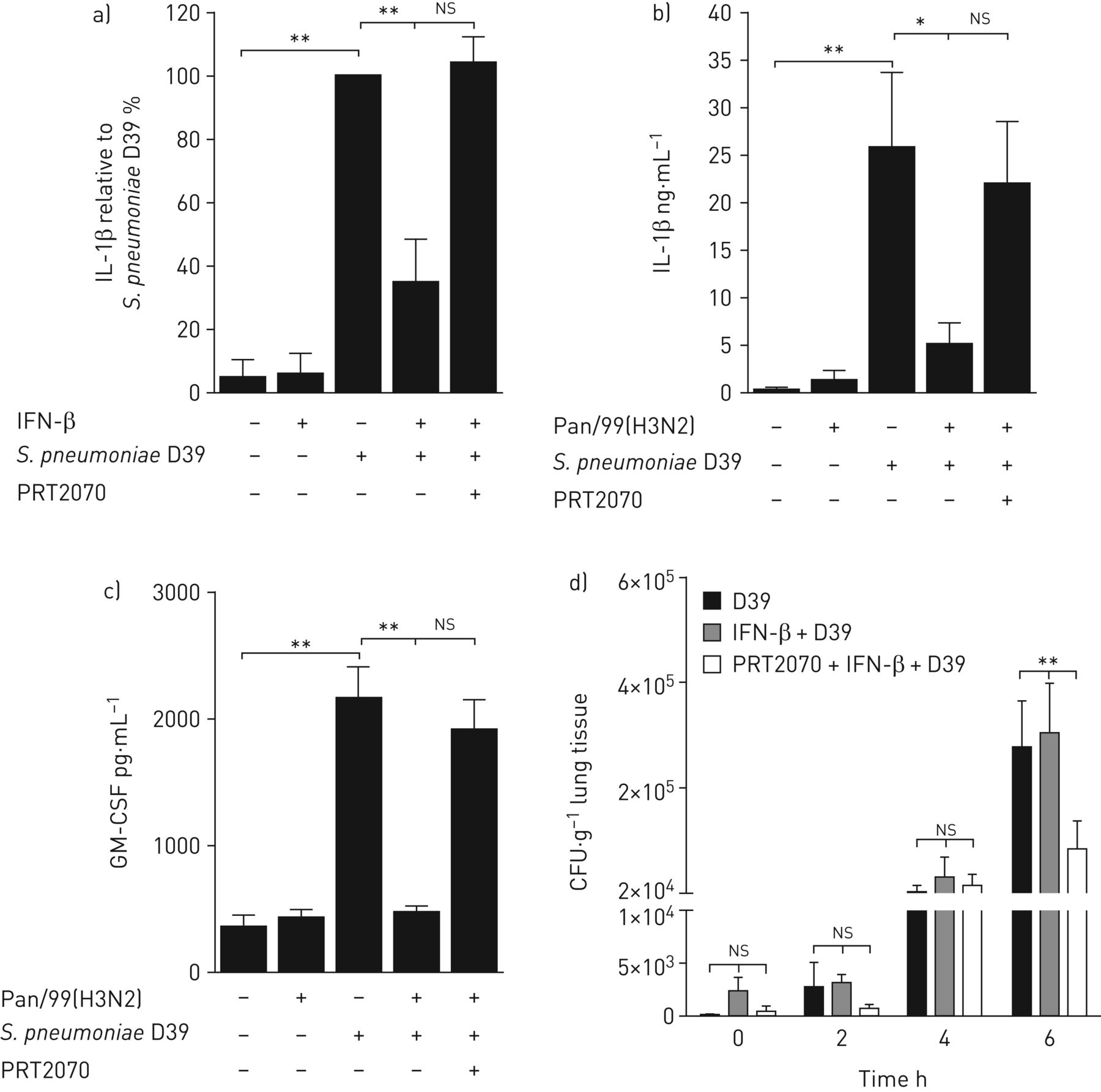
阐明其细胞水平、干扰素类型及作用机制S.肺炎-诱导IL-1β抑制后,我们首先使用从新鲜人肺组织中分离出来的肺泡巨噬细胞,这些巨噬细胞受到细菌的攻击,在不存在或存在I型IFN-β、II型IFN-γ或III型IFN-λ的情况下1.没有差异,对IL-1βmRNA表达的转录水平(实测图4一).然而,I型IFN显著抑制前IL-1β的产生和连续的IL-1β分泌,III型IFN在较低程度上也有抑制作用,而II型IFN在转录和翻译上均无抑制作用(图4 b和c)。与II型IFN相比,IFN型I和III的下游信令包括IFN受体I和III相关的TYK2。因此,我们在肺泡巨噬细胞和人肺中使用了新型可用的小分子竞争性Tyk2抑制剂Prt2070。Tyk2抑制完全恢复IFN-β介导的IL-1β抑制在肺泡巨噬细胞以及IAV诱导的抑制中肺炎链球菌-人肺中触发的IL-1β和GM-CSF生成(图5a- c)。此外,抑制Tyk2有望减少人肺组织中的细菌生长(图5d).
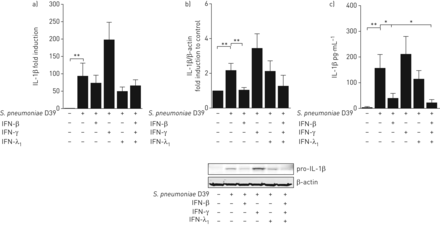
I型干扰素(IFN)-β是肺泡巨噬细胞(AM)诱导的白细胞介素(IL)-1β蛋白表达和分泌的主要抑制剂。来自新鲜人肺组织的AMS,培养2天,并用I型IFN-γ挑战,II II IFN-γ,III型IFN-λ1或所有三个的组合(100 u·ml−1each) for 16 h. Afterwards, cells were infected with肺炎链球菌肺炎料(1)多重感染)。收集RNA、蛋白质和上清液16份 肺炎球菌感染后h,测定a)IL-1βmRNA表达;b)前IL-1β蛋白表达;c)IL-1β分泌的释放。数据以平均值±表示扫描电镜独立实验中至少六个捐献者。*: p≤0.05;**:P≤0.01。
I型和III型干扰素(IFN)信号传导与酪氨酸激酶2抑制剂PRT2070救援肺炎链球菌肺炎料d39诱导肺泡巨噬细胞(AM)和人肺组织中的白细胞介素(IL)-1β和粒细胞-巨噬细胞集落刺激因子(GM-CSF),并降低细菌生长。a)从新鲜人肺组织分离的AMs培养2天,用抑制剂PRT2070(1µM)处理。然后用100 U·mL的IFN-β刺激细胞−1)16小时并感染S.肺炎(1)多重感染;16 h) 测定上清液中IL-1β的释放。b)和c)用PRT2070(1)预处理肺 µM)用于1 h、 所示肺标本用流感病毒Pan/99(H3N2)(IAV)(1×10)激发6 PFU·mL−1), 24小时后用S.肺炎(1×106CFU·毫升−1)再过16年 h、 测量b)IL-1β和c)GM-CSF的释放。a)中的数据以百分比表示S.肺炎-处理过的细胞,因为AM供体之间分泌IL-1β的差异很大。IL-1β分泌量以pg·mL为单位−1被记录在在线补充表S1中。d)术后测量人肺组织中的细菌生长S.肺炎感染,IFN-β处理,接着S.肺炎用PRT2070(1µM)预处理,然后用IFN-β和S.肺炎.数据以平均值±表示扫描电镜至少有五名捐赠者参与了独立实验。ns:不重要。*:p≤0.05;**:p≤0.01.
讨论
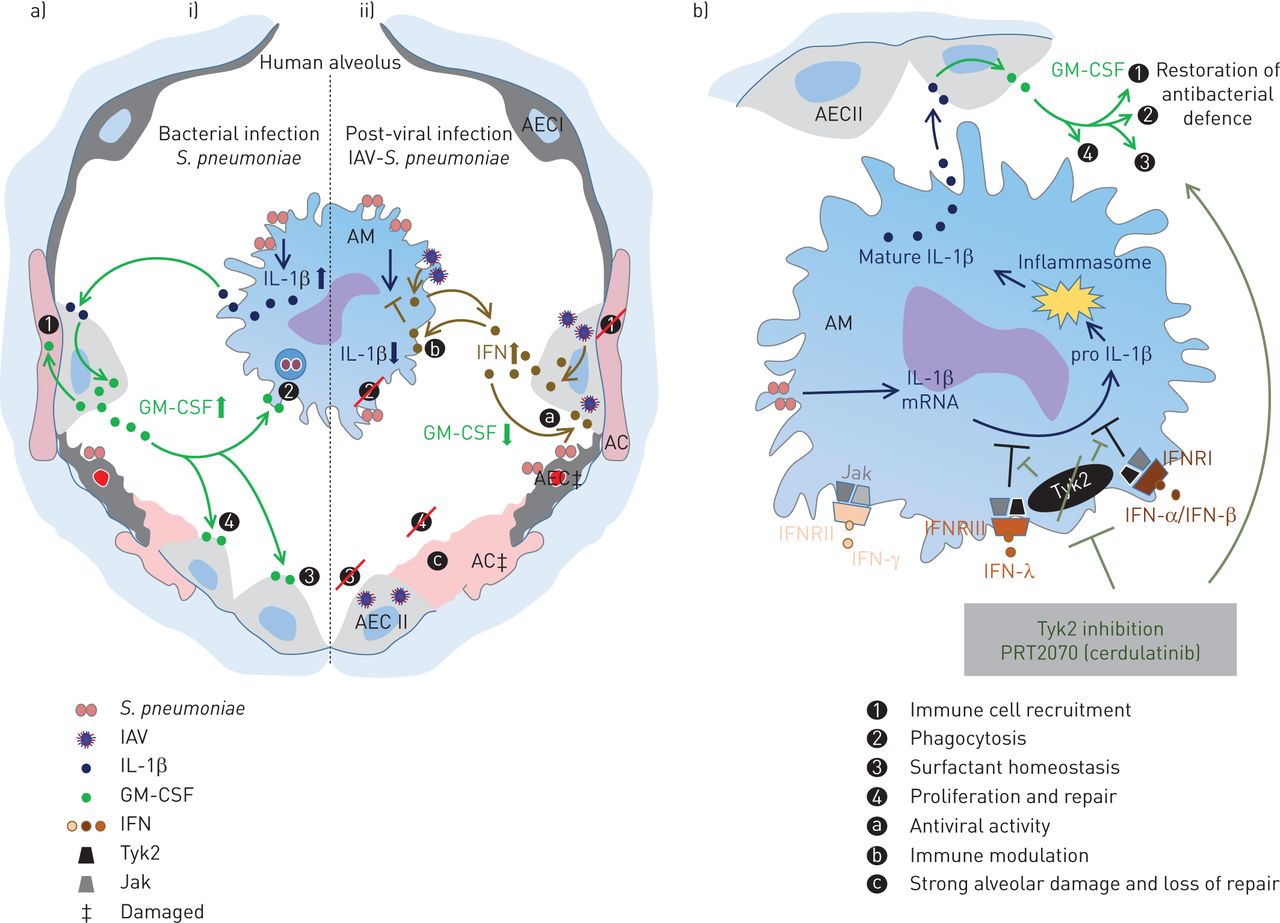
细胞因子的诱导对于启动针对病毒和细菌感染的充分先天免疫反应至关重要。然而,在随后的合并感染中,抗病毒反应可能严重损害抗菌防御,导致细菌杀灭不足、肺泡损伤,最终导致ARDS[4,7]小鼠模型表明病毒诱导的干扰素是调节抗菌作用的重要因素[5- - - - - -7,30.].然而,IFN对人肺组织抗菌免疫应答的影响目前尚不清楚,因此我们使用了an离体人类肺组织模型来解决这个问题。我们发现了肺炎球菌导致肺泡上皮GM-CSF上调的细胞通信通过肺泡巨噬细胞分泌IL-1β,并证明了I型和III IFN产生由AEC II和肺泡巨噬细胞后IAV感染强烈的翻译水平这一机制(干扰图6a).通过IFN受体相关的Tyk2的药理抑制,我们能够完全恢复IL-1β-GM-CSF轴,并对人肺的细菌生长显示有益的影响(图6b).
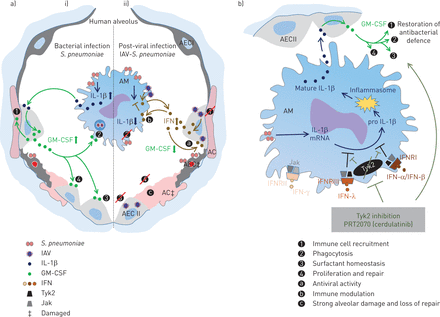
a) 在i)单一细菌或ii)病毒和细菌共同感染下,由人类肺泡中的早期细胞因子反应控制的细胞相互作用的拟议模型肺炎链球菌肺炎料通过肺泡巨噬细胞(AM)识别释放炎症依赖性白细胞介素(IL)-1β,其用作牙槽上皮II型细胞(AECII)中的粒细胞 - 巨噬细胞刺激因子(GM-CSF)产生的驱动因子。GM-CSF可能通过对免疫细胞募集,吞噬功能和表面活性剂稳态的贡献以及增殖和修复过程的贡献来支持炎症反应。II)中,具有流感病毒的预感染病毒引发了抗病毒干扰素(IFN)响应,其随后阻断IL-1β和GM-CSF表达,从而减少了保护性GM-CSF效应并促进肺泡损伤。b)抑制酪氨酸激酶2(Tyk2)与Prt2070(Cerdulatinib)恢复流感病毒平移/ 99(H3N2)(IAV) - 诱导的和IFN型I-和III型辅助抗菌免疫反应的损伤。Jak:Janus Kinases;AC:肺泡毛细血管;IFNR:IFN受体。
在不同物种(如小鼠、雪貂、猪、马和人类)中,连续诱导干扰素是一种保守的、不可或缺的抗病毒反应[31.].虽然细胞抗病毒作用在原则上在物种间似乎相似,但干扰素诱导数百个额外的基因导致多方面的二次免疫反应,我们对其的理解因干扰素类型的物种差异而进一步复杂化,这可能加强了这种多样性[32.].由于除了人类以外,没有物种在IAV感染后会发生继发性细菌性肺炎,看来不同的干扰素效应显著地促成了这种致命的重复感染。这一观点得到了对受IAV毒株感染的小鼠的研究的有力支持,这些毒株易受流感诱导的I型和II型IFN介导的继发性细菌性肺炎的影响[3.- - - - - -7,28.,30.].
因此,我们首先在用季节性IAV菌株H3N2感染后分析了人肺组织中的IFN反应。与鼠标数据相比,显示I类型和主要类型III IFN响应[33.],我们发现人类肺组织显著增加了所有IFN类型的成员,重点是II型IFN-γ,其增加了15倍以上。有趣的是,对A549细胞、原代人类AEC甚至人类肺组织的研究发现了I型和III型IFN的支持结果,但不幸的是忽略了IAV感染后IFN-γ的测量[34.- - - - - -36.].目前的理解是,II型IFN-γ的细胞来源分配给活化的t淋巴细胞和自然杀伤细胞,而不是AECs,因此假设在适应性免疫激活中发挥作用,而不是固有的抗病毒活性[37.].然而,所使用的短期器官培养模型中,适应性免疫反应和活化的T细胞很可能不存在,并且我们鉴定IAV感染AEC II和不肺泡巨噬细胞作为强IFN-γ产生的细胞来源。人类AEC II的产生IFN-γ的能力,是符合对间质性肺病的现有研究[38.].干扰素-γ是否直接在人类肺部发挥抗病毒作用尚不清楚,应在未来的研究中进行研究。在任何情况下,有证据表明,IAV诱导的I型和II型干扰素通过减少中性粒细胞募集、产生IL-17的γδT细胞、趋化因子(如角质形成细胞趋化剂(KC)和巨噬细胞炎性蛋白-2)以及细菌清除,促进小鼠对继发性肺炎球菌感染的易感性[5- - - - - -7].
同样,我们假设IFN受体途径在人类IAV感染中很重要。不幸的是,中性粒细胞或γδ t细胞的招募目前无法测量离体但细胞因子的调节和物种间的比较将提供重要的见解。因此,我们研究了对成分的影响S.肺炎-诱导的细胞因子环境,这是协调适当的先天免疫反应的先决条件。有趣的是,在病毒/细菌共感染期间,细胞因子如IL-6、IL-8、IL-10或TNF-α没有变化,而在小鼠中密斯et al。[28.]表明相同的因子(KC作为IL-8类似物)甚至在共同感染下增加。此外,IL-1β表达和H埃内特et al。[39.]有趣的是,对于人类肺泡,我们发现了一种差异,因为IAV既不增加IL-1β,也不增加GM-CSF,但在肺炎球菌感染下显著抑制了这些因子,这表明了人类肺部共感染发病机制的物种特异性机制。
IL-1β的重要性已在带有IAV和IAV的小鼠共感染模型中得到证实S.金黄色葡萄球菌肺炎球菌肺炎期间,但其影响,特别是IAV感染对人类肺组织的强烈下调,目前尚不清楚[11.,12.]先前的研究表明,IFN可以在不同的水平上抑制IL-1β的产生,这取决于物种或细胞类型[8- - - - - -10.,40,41.]因此,我们用IFN-β和IFN-γ预处理取代IAV,并发现类似的表达模式,在人肺中IL-1β和GM-CSF显著减少。
除了IL-1β,一些研究已经证明GM-CSF在IAV肺部感染或肺炎球菌性肺炎的预后中具有相当大的作用,并且已经引入了第一个使用GM-CSF作为辅助治疗的临床试验[15.- - - - - -17.,19.- - - - - -23.,42.,43.].这强调了直接在人肺组织中阐明其调节的重要性,特别是在病毒/细菌的共感染,其中解毒的免疫反应可能会显着促进致命结果。
GM-CSF可能由不同的刺激物诱导,在小鼠肺中Cakarovaet al。[29.]发现TNF-α是脂多糖刺激后的负责介质。然而,小鼠AEC中GM-CSF的调节并不固定于TNF-α,似乎取决于细胞来源和培养条件,决定了表型特征ir -K阿西莫夫et al。[44.]已经证明GM-CSF在在体外小鼠AEC可由TNF-α和IL-1β诱导,证明了在人类环境中验证此类结果的重要性。在人类肺部,细胞来源以及驱动因素目前尚不清楚。肺炎球菌Toll样受体的直接激活或继发性细胞因子介导的效应可能导致GM-CSF的表达。我们的实验在完整的人肺组织、分离的原发性AEC和肺泡巨噬细胞(使用Anakinara作为特异性IL-1受体拮抗剂)上的实验表明,肺泡巨噬细胞衍生的IL-1β-而非TNF-α促进AEC II依赖性GM-CSF诱导的间接肺炎球菌机制。我们的观察进一步证实了这一点,即在合并感染的情况下,TNF-α仍然存在,单独刺激TNF-α不足以诱导GM-CSF。
与GM-CSF的诱导相比,它对AEC II的抑制也特别有趣,因为ifn已被证明直接抑制IL-1β -诱导的GM-CSF在人视网膜上皮细胞或骨髓细胞中[13.,45.].因此,我们证明,除了IL-1受体阻断外,IFN在IL-1β存在的情况下抑制了肺GM-CSF,这意味着IFN既作用于肺泡巨噬细胞IL-1β的产生,也作用于IL-1β诱导的继发性上皮GM-CSF表达。
我们的研究结果表明,在人肺泡中有一种独特的细胞通讯机制,肺泡巨噬细胞被激活S.肺炎产生IL-1β,然后触发上皮GM-CSF的产生。这种信号级联被先前的IAV感染所抑制。GM-CSF再一次被证明在肺部感染中起关键作用[15.- - - - - -21.]现在被认为是急性肺损伤和ARDS的治疗[22.,23.].因此,我们的目的是找到一种免疫调节干预策略,以恢复抗菌GM-CSF响应。进行系统性分析以显示负责IFN类型以及IL-1β抑制的细胞水平,发现I型和III IFN块亲IL-1β的翻译。与I型IFN的小鼠的治疗显示出导致增加IAV诱导的死亡率是由于强的促炎免疫细胞的激活[46.].与II型IFN受体相比,I型和III受体信号中的下层溶解于TYK2,已经显示在鼠巨噬细胞中的信号传感器和转录1依赖性方式中的IL-1β表达进行控制[47.].患有Tyk2的几种临床试验在患有免疫学和炎症疾病的患者中正在进行中48.],但目前还不清楚这是否干预可能是肺部感染有帮助,特别是在严重的病毒/细菌性肺炎。在这里,我们表明,使用的药理学的Tyk2抑制剂(PRT2070)完全恢复肺炎球菌诱导的肺泡巨噬细胞IL-1β,并最后上皮GM-CSF IAV感染人肺癌共感染后。这对于临床试验在肺炎相关ARDS接收GM-CSF治疗的患者特别感兴趣的。在以P试用全身用药皇家艺术et al。[23.]没有揭示显着的影响,而是一个第一个富有同情心的吸入应用,患者数量较少eroldet al。[22.]表现出肺宿主防御调节的积极趋势。如果GM-CSF治疗成功,与IFN受体途径抑制相比,不良反应可能更少。然而,如果单一因素如GM-CSF不足以恢复整个肺宿主对继发性细菌性肺炎的防御,例如,临床开发的Tyk2抑制剂,早期IFN受体通路调节将提供一个更有前景的方法,因为我们不仅恢复内源性GM-CSF,同时也证明了Tyk2抑制对减少细菌生长的有益作用。
综上所述,我们的结果首次证明人类肺组织模型适合描述IAV和IAV中的先天免疫事件S.肺炎合并感染。由于缺乏系统性白细胞募集和适应性免疫,区分肺泡特异性调节成为可能,以模拟早期细胞因子控制的肺泡巨噬细胞和AEC之间潜在的细胞相互作用。
必须强调的是,大多数组织来自有吸烟史和慢性阻塞性肺疾病(COPD)的肺癌患者作为一种共病。因此,所使用的周围正常肺组织不能被视为完全健康。然而,吸烟是社区获得性肺炎(CAP)的主要危险因素,COPD患者发生CAP和严重IAV相关肺炎球菌感染的风险高出两到四倍[49.- - - - - -51.].这反映在大型CAP队列研究中,表明四分之一到三分之一的住院患者患有慢性阻塞性肺病[52.].因此,我们不会归类吸烟作为模型的限制,而是作为一个非常相关的方面,我们认为,我们从他们那里获得的组织中的患者是在CAP的风险较高,可以认为是利益相关群体。
与小鼠模型相比,人类肺组织的另一个好处是,人类肺组织可以直接感染从受感染患者分离的病毒和细菌菌株,因此加强了数据的相关性。如本研究所示,揭示小鼠和人类之间的免疫差异尤其重要ar的重要性,因为GM-CSF等因素已经用于临床试验,并被考虑用于急性肺损伤的免疫调节治疗。此外,我们建议将药物性Tyk2抑制作为一种免疫调节干预策略,以维持严重病毒/细菌性肺炎中关键的内源性GM-CSF水平。
补充材料
补充材料
请注意:补充材料不是由编辑部编辑的,而是由作者提供的。
补充材料erj - 01953 - 2016 - _supplement
图S1。甲型流感病毒Pan/99(H3N2) (IAV)和肺炎链球菌肺炎料D39(S.肺炎)在单一和共同感染的人肺组织中以及在单一和共同感染的人肺组织中IAV诱导的干扰素(IFN)表达。(A)人肺组织被模拟感染,(B)用季节性IAV激发24小时,(C)S.肺炎16小时,或(d)随后与IAV进行共感染S.肺炎.IAV(绿色、白色箭头)通常在pro-SP-C(蓝色、星号、B、D)指示的肺泡上皮II型细胞中复制。S.肺炎与肺泡上皮细胞(开放箭头,C,D)和肺泡巨噬细胞(白色箭头,D)紧密相连。通过差异干扰对比(灰色)观察肺结构,并使用DAPI(橙色)对细胞核进行复染。比例尺为10µm。ERJ-01953-2016\u补充图\u S1
图S2甲型流感病毒Pan/99(H3N2)(IAV)和肺炎链球菌肺炎料(S.肺炎)显示不同的细胞因子调节。肺外植体离体模拟感染或挑战IAV (1 × 10)6(A-F)之后,将专用标本感染S.肺炎(1 × 106CFU/ml) (A - D)或(E, F)血清3型临床分离株S.肺炎(1 × 106分别CFU /毫升)。肺炎球菌感染后16小时收集上清,并检测(A) IL-6, (B) IL-8, (C) IL-10, (D) TNFα, (E) IL-1β, (F) GM-CSF的释放情况。在独立实验中,6个供体的数据以平均值±SEM表示。* * p≤0.01。ERJ-01953-2016\u补充图\u S2
图S3。甲型流感病毒Pan/99(H3N2) (IAV)感染或干扰素(IFN)治疗对人肺组织未能抑制肺炎链球菌肺炎料D39(S.肺炎)诱导肿瘤坏死因子α(TNFα)释放离体甲型流感病毒Pan/99(H3N2) (IAV) (1 × 10)模拟感染或攻毒24 h6PFU/ml)或用IFNβ和IFNγ组合(各100 U/ml)预处理16小时。模拟或S.肺炎(1 × 106CFU / ml)在另外16小时后,分析肺样本的上清液,以释放TNFα。在独立实验中,6个供体的数据以平均值±SEM表示。* * p≤0.01。ERJ-01953-2016\u补充图\u S3
图S4。TNFα刺激诱导环氧合酶-2(COX-2)表达和粒细胞巨噬 - 集落刺激因子(GM-CSF)的表达是时间移位到IL-1β在人体肺部。(A)刺激人肺标本16小时诱导促炎性COX-2的TNFα(100纳克/毫升)。(B)人肺外植要么模拟感染或用激发S.肺炎(1 × 106CFU/ml) 2、4、6、8和16 h,显示GM-CSF的时移释放与早期IL-1β释放有关。数据显示为对照的fold和独立实验中3 / 4个供体的平均值±SEM。ERJ-01953-2016\u补充图\u S4
图S5.从新鲜人肺组织中分离肺泡巨噬细胞(AM)和肺泡上皮II型细胞(AEC II),并将其接种在玻璃盖玻片上,通过免疫荧光和共焦显微镜用不同的细胞标记物固定和显示。(A)培养2天的AM对CD-68(红色)呈阳性.AEC II在分离后培养4天。(B)细胞泛细胞角蛋白(红色)和前SP-C(绿色)呈阳性。核复染使用DAPI(蓝色)。显微镜:卡尔蔡司LSM780,Plan Apo Chromat 63×oil/NA 1.4。标尺5µmERJ-01953-2016\u补充图\u S5
图S6。干扰素(IFN)未抑制人肺泡巨噬细胞(AM)肿瘤坏死因子α (TNFα)的表达。分离的AM培养2天,在肺炎球菌感染前,用干扰素β和γ联合(100 U/ml)刺激或模拟攻击16小时。然后,检测上清液中TNFα的释放情况。在独立实验中,4位供体的数据以平均值±SEM表示。* p≤0.05。ERJ-01953-2016_补充图_S6
图S7.干扰素(IFN)抑制人肺组织中IL-1β诱导的粒细胞-巨噬细胞集落刺激因子(GM-CSF)释放。用IFNβ和IFNγ组合(各100 U/ml)预处理人肺16 h,随后进行IL-1β刺激(10 ng/ml)16小时。分析肺标本上清液中GM-CSF的释放。数据以独立实验中三个供体的平均值±SEM表示。*p≤0.05.ERJ-01953-2016\u补充图\u S7
披露
补充材料
d . Fatykhovaerj - 01953 - 2016 - _fatykhova
A.C.霍克erj - 01953 - 2016 - _hocke
K. Zscheppang.ERJ-01953-2016zscheppang
致谢
这项工作的一部分包含在Johanna Berg的博士论文中。我们感谢Katharina Hellwig和Doris Stoll(Charité-Universitätmedizin Berlin,Berlin,Germany)提供的技术援助。作者感谢Thorsten Wolff(Robert Koch Institut,Berlin,Germany)提供的甲型流感病毒H3N2。肺炎链球菌肺炎料D39血清型2由Sven Hammerschmidt(Greifswald,Greifswald,Greifswald,Greifswald,Greifswald,临床分离型3,由Mark Van der Linden(德国亚琛(Aachen)的Streptocooci国家参考中心)友好提供。
脚注
本文提供了补充材料www.qdcxjkg.com
支持声明:该研究由德国教育和研究部(BMBF - PROGRESS)资助给A.C. Hocke (C8),由德国研究基金会(DFG SFB-TR84)资助给N. Suttorp (B1), A.D. Gruber (Z1b), A.C. Hocke (Z1a)以及A.C. Hocke和S. Hippenstiel (B6)。本文的资金信息已存入交叉参考基金登记处.
利益冲突:可以在本文旁边找到披露www.qdcxjkg.com
- 收到2016年10月5日。
- 接受2017年4月7日。
- 版权所有©ERS 2017











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}