





摘要
尽管现代药剂疗法,肺动脉高压仍然是致命疾病。骨形态发生蛋白受体II型基因中的突变(BMPR2)导致BMPR2表达减少,这与PAH有因果关系。BMPR2主要表达于肺内皮细胞,并与转化生长因子(TGF)-β信号转导机制相互作用。
我们的目标是评估上调的PAH的效果BMPR2通过针对腺嘌呤BMPR2递送到肺血管内皮的基因。我们使用了两种成熟的PAH:慢性缺氧和六公民石斛(MCT)。
在这两种高血压模型,那些接受BMPR2较低的心室肥大,肺血管阻力较少,心肌功能改善,血管重塑减少。在MCT模型中,TGF-β增加,这是预防的BMPR2治疗。体外, TGF-β1诱导人肺微血管内皮细胞内皮-间充质转化(endomt),与BMPR2表达降低相关。即使在TGF-β1持续存在的情况下,通过适当的配体刺激BMPR2信号,EndMT也可以部分改善。
综上所述,这些结果表明了该基因上调的治疗潜力BMPR2轴在PAH,其可以部分地由打击TGF-β的影响重塑介导。
肺动脉高压(PAH)是一种致命的致命疾病,其特征在于肺血管改造,包括血管内皮细胞的异常增殖,肌肉肥大平滑和内膜增稠,因此肺血管阻力增加了[1,2GydF4y2Ba].该疾病导致进步呼吸困难和右心力衰竭。骨形态发生蛋白受体II型基因中的突变(BMPR2)与多环芳烃有因果关系[3.,4].已发现突变存在于患有成本疗法PAH的“散发性”病例的70%和〜20%的“散发性”病例中,并导致BMPR2表达和信号传导[5].BMPR2是转化生长因子(TGF)-β超家族受体的成员。有证据表明,PAH发病机制涉及无序TGF-β信号,可能的话,BMPR2和TGF-β信号传导机制之间的串扰。BMPR2表达主要见于肺血管内皮,虽然血管平滑肌表达也存在。减少BMPR2表达也被在患有继发PAH肺血管观察,暗示该途径也可能是多种常见的临床情况下重要的发病机制,超出那些与BMPR2肺动脉高压是一个特征的突变[6].尽管药理学治疗的进步导致了PAH患者的改善结果[7]最近来自法国和美国的国家注册表数据继续表现出这种疾病的不可接受的高死亡率,表明需要进一步治疗的需求[8- - - - - -10.].
在PAH的研究中广泛使用了各种动物模型,尤其是慢性缺氧和野百合碱(MCT)大鼠模型[11.].这些模型中最近的研究表明,BMPR2水平降低和异常骨形态发生蛋白(BMP)信号传播[12.,13.].我们以前表现出预处理BMPR2基因递送降低了缺氧攻击的大鼠的PAH反应[14.但对已确定的多环芳烃的影响尚不清楚。BMPR2导致PAH的机制尚不清楚,但该信号通路对细胞增殖和分化有许多影响。BMPR2和TGF-β信号通路之间存在复杂的串扰[15.].值得注意的是,虽然这些动物模型中BMPR2表达降低,但TGF-β表达增加[16.,17.].
在目前的研究中,我们首先寻求判断BMPR2途径是否可以被利用作为既定疾病的潜在治疗目标,IE。独立刺激,慢性缺氧和MCT。我们利用我们以前开发的腺病毒(AD)载体肺靶向策略,其中我们将AD载体与双特异性抗体缀合物联系起来,该抗体缀合物靶向血管紧张素转换酶(ACE),膜结合的蛋白酶高度表达肺内型细胞[18.,19.].我们已经表明这种方法改善了肺内皮基因递送[20.,21.].
在此,我们显示交付BMPR2:1)改善了缺氧或MCT挑战症诱导的BMPR2表达的下降;2)改善了MCT模型中所见的TGF-β表达的增加;3)减少肺动脉高压和相关血管重塑的发育。体外,我们发现BMPR2信号可能通过改善TGF-β诱导的内皮形态变化和内皮-间充质转化(EndMT)而起作用。
方法
动物与实验设计
所有动物方案进行了审查,并通过医学和兽医学科学研究所动物研究伦理委员会(IMVS)(皇家阿德莱德医院和阿德莱德大学,阿德莱德,SA,澳大利亚)的批准。成年雄性SD大鼠(6-7周龄,250-350克的机身质量)使用。大鼠被安置在IMVS动物护理设施,并输送食物和水自由.使用了两种型号,缺氧和MCT诱导。对于缺氧,在我们先前描述的源极缺氧室中保持大鼠[14.](10%氧气)3周,然后分配到两种治疗缺氧基团。然后给予尾静脉注射Adtrackluc(无关病毒对照)或ADBMPR2,每个抗病毒/抗ACE双特异性偶联缀合物(Fab-9b9)预孵育[14.].然后将大鼠送回缺氧室3周,然后通过心导管检查肺动脉高压。对于MCT,将大鼠分为三组(两个MCT组(60 mg·kg))−1,皮下注射)和一个盐对照组)。MCT后10天,用Adtrackluc + Fab-9b9或Adbmpr2 + Fab-9b9注射大鼠。经过进一步的8-10天,PAH被评估。对于MCT和病毒注射,大鼠短暂麻醉(异氟烷〜5分钟)。每次剂量广告矢量(1×1010.在室温下与6 μg Fab-9B9复合物30分钟后注射。注射通过采用27G胰岛素注射器(Terumo, Elkton, MD, USA),用无菌PBS将Ad总量带入250 μL。
请参阅有关的病毒构建信息,双特异性结合,血流动力学测量,免疫组化和免疫印迹网上补充。
细胞培养
Clonetics®肺衍生的正常人体肺部微血管内皮细胞(HPMVECS)(Cambrex Bio Sckets Inc.,Walkersville,MD,USA)在EGM®-2MV(Cambrex Bio Science Inc.)培养,补充有5%胎牛如供应商的推荐,血清,并且在段落4和9之间使用。所有细胞都在5%的CO中生长2GydF4y2Ba气氛在37°C。
诱导EndMT
在HPMVEC单层中评估TGF-β1诱导Endmt的能力。电池被镀(3×105每井;~50-60%汇合)进入六孔板和24小时后,培养基补充重组人TGF-β1(5 ng·mL−1;研发系统,明尼阿波利斯,MN,美国)诱导EndMT。培养物保持在5% CO的湿润环境中2GydF4y2Ba在37℃下培养箱,所有培养基发生每48小时,细胞生长21天。在1:10比例下,未经暗示的HPMVEC每周传代,而TGF-β-处理的细胞以1:5的比例每周传代两次,以保持细胞活力。通过丧失血管内皮(Ve) - Cadherin的丧失和间充质标记物的增益评估Endmt(细胞外结构域的细胞纤维纤维蛋白的剪接变体)和S100A4的增益。采用奥林巴斯CKX41显微镜进行相位对比显微镜图片,配备奥林巴斯DP20相机(奥林巴斯,日本Olympus)。
Endmt逆转实验
TGF-β1培养18天后,用含TGF-β1和重组人(rh)BMP-2 (333 ng·mL)的新鲜培养基替代培养基−1;R&d系统)或的rhBMP-7(325纳克·毫升−1;研发系统)。媒体再次在48小时内再次发生变化,实验后24小时停止。在线补充,提供了用于免疫荧光检测和TGF-β-诱导的Endmt的免疫荧光检测和免疫斑分析的方法。
统计分析
数据以平均值±表示SEM..组间比较的装置使用双尾非配对t检验或单向ANOVA,在适当情况下执行的。当ANOVA F-值指示的显著差异,学生纽曼-Keuls多重比较检验。然后用(SIGMASTAT; SYSTAT软件公司,点里士满,CA,USA)。的<0.05的p值被认为是统计学显著。
结果
肺血管BMPR2-myc的免疫检测
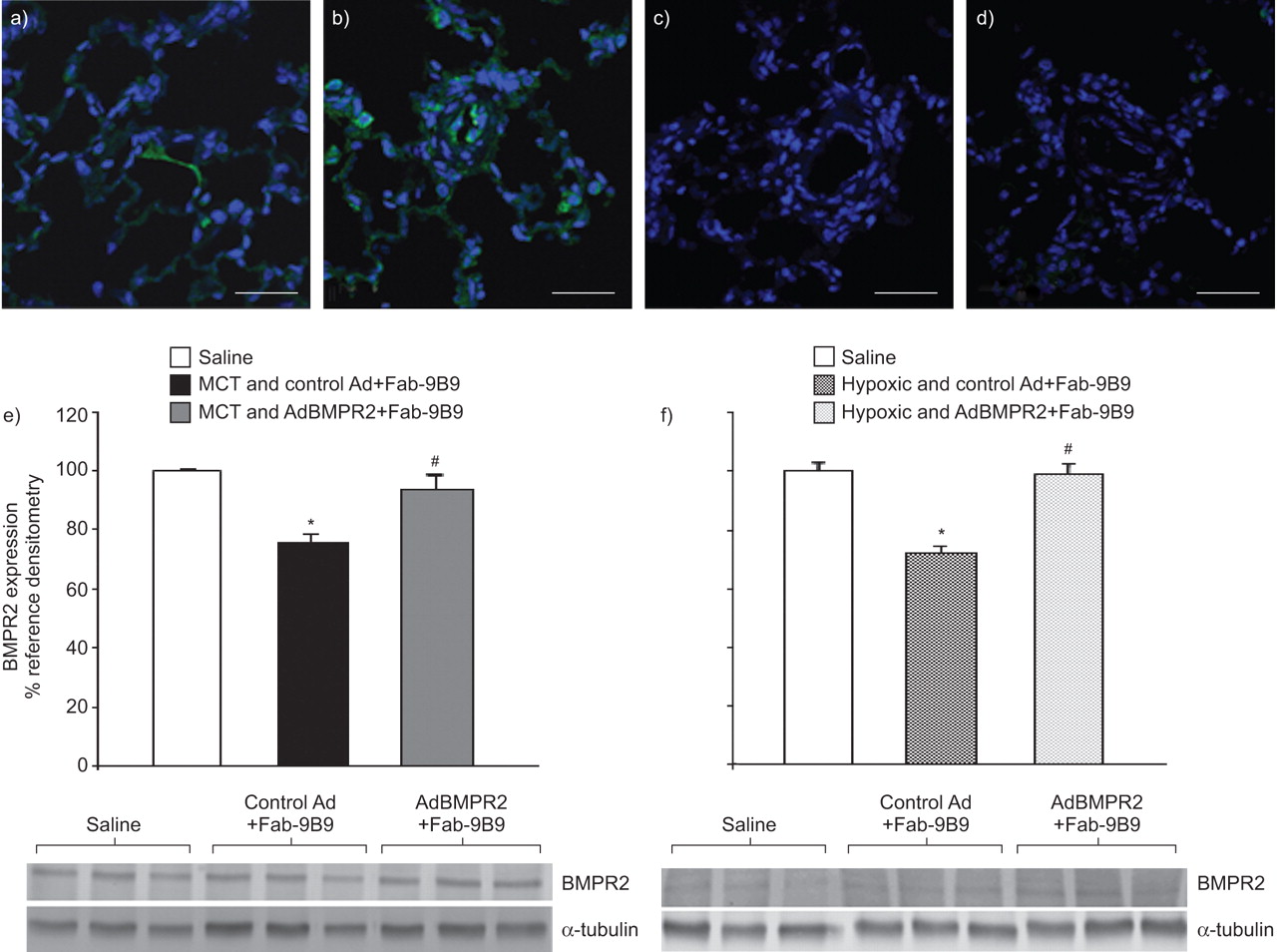
肺靶向AD介导BMPR2-myc基因通过MYC标签的免疫荧光染色证实了MCT处理的大鼠中的转基因表达。MCT注射后接受ADCMVBMPR2MYC + FAB-9B9 10天的大鼠,然后在ADBMPR2尾静脉注射后3天杀死3天,在小型肺血管和肺泡毛细血管中显示出正绿色染色,确认BMPR2-myc基因转基因表达(图1一个和b)。这些结果都符合我们的使用该载体和此前公布的数据使用报告基因[类似的研究一致20.,21.].无一抗孵育的切片未见染色(无花果。1C.)或只注射mct的大鼠抗c-myc染色切片(无花果。1D).心脏组织切片未见染色。
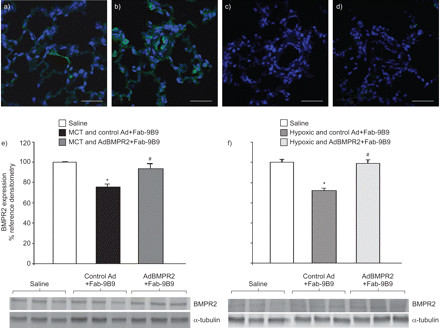
免疫荧光(绿色)检测肺血管中骨形态发生蛋白受体II型(BMPR) -myc。a-c) monocrotaline (MCT)处理的大鼠肺注射腺病毒(Ad)CMVBMPR2myc与抗病毒/抗血管紧张素转换酶双特异性抗体偶联物(Fab-9B9)预孵育。a, b)切片进行抗c-myc染色。c)无初级抗c-myc的切片。d)仅经mct处理的大鼠抗c-myc染色。比例尺= 50 μm。e) MCT-和f)缺氧处理的大鼠中,通过免疫印迹图像分析定量BMPR2表达。BMPR2和α-微管蛋白表达的代表性免疫印迹分别显示在它们各自的图下面。数值计算为相应BMPR2信号在管家蛋白(α-微管蛋白)上的化学发光比例,然后用与生理盐水组相比的比例变化百分比表示。数据以平均值±表示SEM..N = 6只动物每个治疗组中使用。*:P <0.05相对盐水;#:P <0.05处理和控制AD + FAB-9B9相对治疗和ADBMPR2 + FAB-9B9。
与生理盐水注射的大鼠相比,暴露于MCT和慢性缺氧的大鼠肺BMPR2表达显著降低,这是通过福尔马林固定石蜡包埋(FFPE)组织切片提取的蛋白免疫印迹检测到的(图1 e和f,分别地)(平均值±SEM.BMPR2:表达盐水相对未经中华人民共和国交通部+控制广告+ Fab-9B9 100.0±1.4%相对75.6±2.9%(P <0.05);盐水相对慢性缺氧+对照组Ad+Fab-9B9 100.0±7.8%相对72.0±5.5%(P <0.05))。重要的是BMPR2Ad载体靶向策略可纠正MCT和缺氧诱导的BMPR2表达缺陷。在腺病毒感染后10-21天观察效果BMPR2基因传递。
广告载体对肺血流动力学中MCT和缺氧诱导变化的影响
在麻醉和手术干预(MCT和慢性缺氧;表1).MCT和慢性缺氧诱导显着的PAH。右心室收缩压(RVSP)和平均肺动脉压(P̄.巴勒斯坦权力机构通过每项研究的终点显着增加,在MCT后更明显的增加(无花果。2A和b,和表1).两种模型PAH诱导心脏函数的显着降低,如下降的心输出(CO)和心脏指数(CI)(无花果。2C和d,表1(分别)和显着增加肺电阻指数(肺血管阻力指数(PVRI)和总肺血管阻力(TPVR))(无花果。2e和f,表1, 分别)。
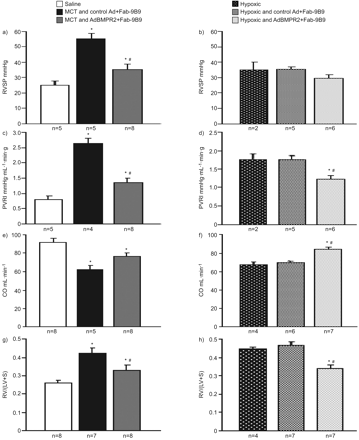
腺病毒(AD)BMPR2给药对A,C,E,G)偏霉素(MCT)和B,D,F,H)慢性缺氧的进一步发展肺动脉高压进一步发展的影响。a,b)右心室收缩压(RVSP)A)MCT和B)缺氧;C,D)C)MCT和D)肺血管抗性指数(PVRI)缺氧;E,F)e)中的心输出(CO)MCT和F)缺氧;和G,H)富尔顿指数(右心室(RV)与左心室(LV)+隔膜的比率(LV)+隔膜),MCT和H)缺氧。数据以平均值±SEM..*:P <0.05相对控制;#:P <0.05处理和控制AD + FAB-9B9相对治疗和ADBMPR2 + FAB-9B9。
在MCT研究中,与接受无关病毒控制(Adtrackluc + Fab-9b9)的MCT处理的大鼠相比,那些接受BMPR2(ADBMPR2 + FAB-9B9)显着降低了PAH响应(RVSP比下降36%P̄.巴勒斯坦权力机构在30%以下)和血管阻力(TPVR为38%以下和PVRI率为48%降低),和显著改善心脏功能(CO是高22%和CI为25%以上)(无花果。2A, c和e表1).在慢性缺氧研究中看到了类似的结果,IE。与接受对照载体的缺氧动物相比,肺靶向ADBMPR2处理的动物显着降低了TPVR(较少的29%)和PVRI(较少30%),共同和Ci的改善20%。虽然BMPR2慢性缺氧治疗降低了RVSP和P̄.巴勒斯坦权力机构,该数值并没有在这种情况下达到统计学意义(P = 0.07)(无花果。2B.,d和f,和表1),表明这些指数的测量对变化不太敏感。
在MCT和慢性缺氧暴露之后,观察到严重的右心室肥大(评估为右心室(RV)游离壁重量与隔膜和左心室(LV)游离壁重量的总和)(无花果。2G和h)。在慢性缺氧的研究中,肥大的程度并没有在对照Ad +的Fab-9B9处理的动物(改进的平均值±SEM.RV./ (LV + S.)0.471±0.02),而单独缺氧(0.445±0.02)。在MCT和缺氧研究中,RV/ (LV + S.)通过目标显着降低BMPR2基因传递(AdBMPR2+Fab-9B9)与各自的对照大鼠(慢性缺氧对照Ad+Fab-9B9)进行比较相对ADBMPR2 + FAB-9B9 0.471±0.020相对0.341±0.02(P <0.05);MCT控制AD + FAB-9B9相对AdBMPR2 + Fab-9B9 0.425±0.03相对0.333±0.02(P <0.05))。在MCT和缺氧的研究都在所有治疗组中检测到LV(包括S)无显著差异。在MCT的研究中,LV + S在生理盐水,MCT对照Ad +的Fab-9B9-和MCT AdBMPR2 +的Fab-9B9处理的大鼠为0.735±0.033,0.697±0.027和0.696±0.023克,分别,而在慢性缺氧,缺氧对照Ad +的Fab-9B9和缺氧AdBMPR2 +的Fab-9B9,LV + S为0.747±0.029,0.742±0.021分别和0.821±0.031克。
对血管肌肉发酵的影响
五个大功率肺野在从每个治疗组,其中〜50微米直径的完全muscularised容器的数量进行计数,四到五只大鼠进行了研究。如图所示图3A,MCT与盐水相比,肌肉大小的血管数量诱导显着三倍(44±8相对分别为15±1.5;p < 0.005)。有针对性的BMPR2基因传递显著降低了mct诱导的血管肌肉化增加42%。在慢性缺氧暴露后,观察到大量完全肌肉化的血管,而对照组Ad给药组(缺氧)的血管数量没有变化相对控制AD + FAB-9B9 49±3.2相对57±2.4),而靶向ADBMPR2治疗显着降低了完全肌肉大小的血管的数量至31±1.9(P <0.05)(图3 c).在MCT和缺氧实验中,平滑肌区域也有类似的效果(图3 b和d,分别地)。未观察到平滑肌区域A显著减少40%以下的目标BMPR2与相应的对照AD + FAB-9B9组相比。代表每个治疗组的肺组织切片和用于平滑肌α-肌动蛋白的免疫染色图3E-J.
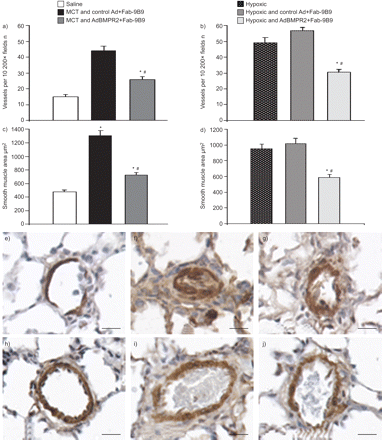
小(~50μm)的数量每10种随机大功率场(200×)的全肌肉大小,通过平滑肌α-肌动蛋白染色中的一个半甲酰胺(MCT)和B)缺氧研究。平均肌肉面积为10 50微米血管C)MCT和D)缺氧研究。数据以平均值±表示SEM.(每组4-5只)。*:P <0.05相对控制;#:P <0.05处理和对照腺病毒(AD)预孵育与抗病毒/抗血管紧张素转换酶双特异性抗体缀合物(FAB-9B9)相对治疗和AdBMPR2 + FAB + 9B9。基于E小动脉)盐水对照,F)MCT和对照Ad + Fab9B9,克)MCT和AdBMPR2 + Fab9B9,h)的缺氧,i)的缺氧和对照Ad + Fab9B9,和j的代表高功率显微照片)缺氧和AdBMPR2 + Fab9B9动物。切片进行平滑肌α肌动蛋白。比例尺=50μm以下。
对细胞增殖的影响
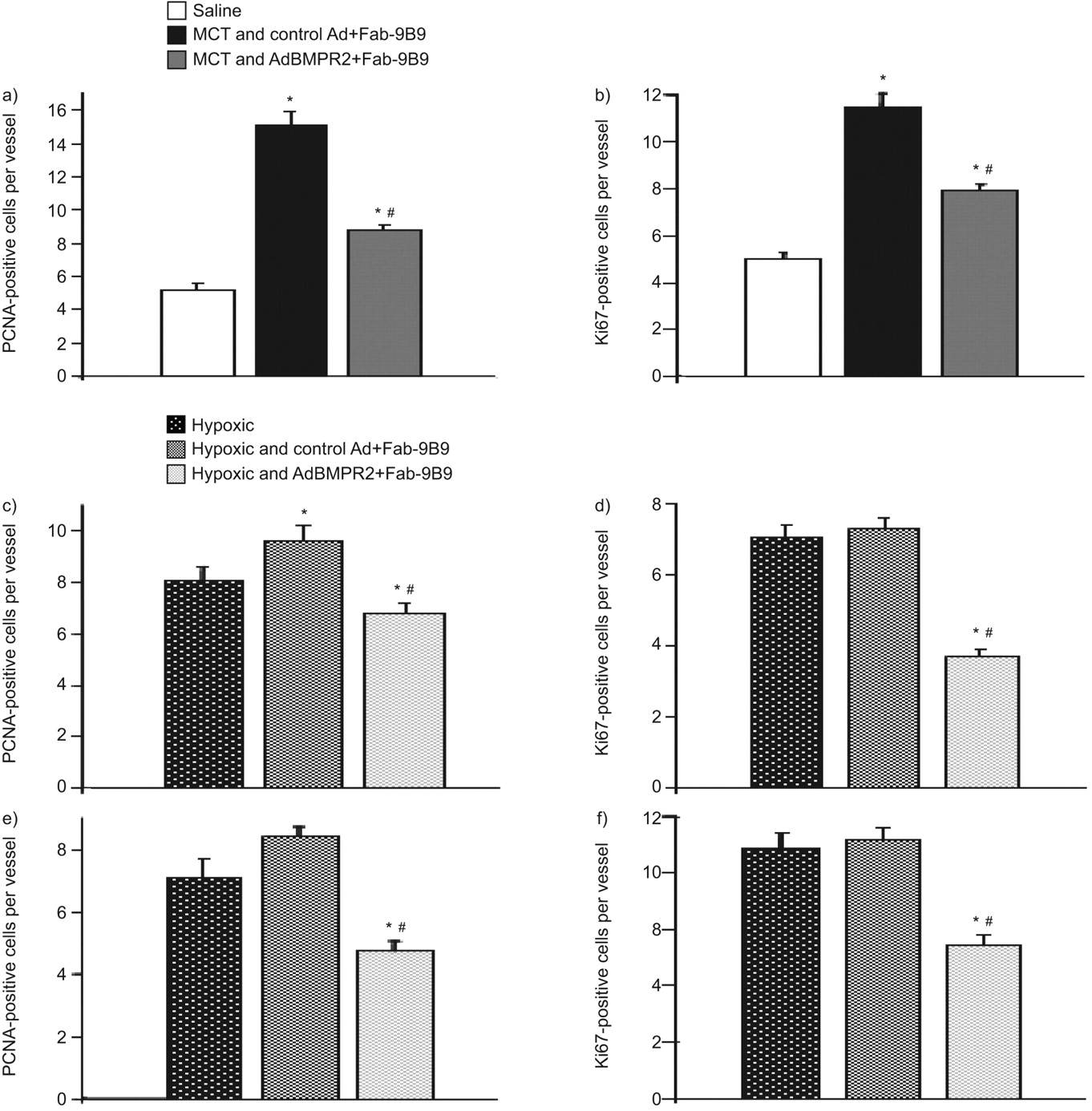
我们进一步评估了增殖细胞核抗原(PCNA)和Ki67的切片,发现盐处理大鼠肺中很少有明显的增殖细胞;然而,MCT给药3周后,内皮病变和靶区细胞增殖增加BMPR2基因传递降低了增殖反应(图4和B分别)。在暴露于慢性缺氧动物,增加的PCNA和Ki67染色在内皮细胞和平滑肌细胞中看到;控制广告投放并没有改变这种增殖反应,而有针对性BMPR2PCNA-和ki67阳性内皮细胞(图4摄氏度和d)和平滑肌(图4 e和f分别)细胞数。
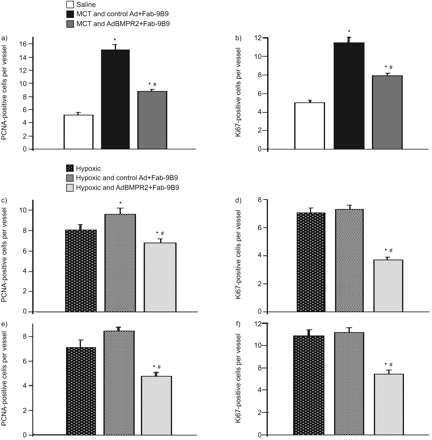
A,C,E)增殖细胞核抗原(PCNA)的量化 - 和b,d,f)在一个Ki67的阳性细胞核,B)野百合碱(MCT)诱导的小(〜50微米)的血管内皮细胞损伤,C,d)内皮细胞和E,F)的血管平滑血管的小(〜50微米肌肉细胞)在缺氧处理组。每只动物10个容器进行了评估。N = 4-5只动物每组。数据以平均值±SEM..*:P <0.05相对控制;#:P <0.05处理组和对照腺病毒(AD):+的Fab-9B9相对治疗和ADBMPR2 + FAB-9B9。
对肺TGF-β表达的影响
免疫印迹法检测FFPE切片TGF-β, mct处理大鼠TGF-β水平显著升高(平均±)SEM.100.0±20.1%相对生理盐水组172.6±26.1%相对MCT和控制AD + FAB-9B9分别;无花果。5A).AdBMPR2治疗减弱了这种反应(121.7±24.3%)。与此分析一致的是,我们发现,在接受靶向治疗的动物中,重塑血管内皮病变中TGF-β免疫染色增加BMPR2送货 (图5罪犯).缺氧组大鼠TGF-β表达差异无统计学意义(100.0±23.4%)相对盐水100.7±43.2%相对而在低氧和AdBMPR2+Fab-9B9组,TGF-β表达量为103.3±22.7%)。其他人在mRNA水平上也观察到了类似的结果[17.].
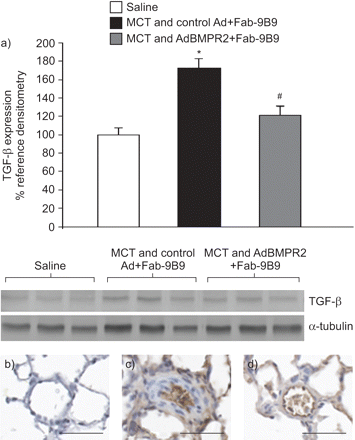
用抗病毒/抗血管紧张素转换酶双特异性抗体缀合物(Fab-9b9)预孵育的盐水,偏甲胺(MCT)和对照腺病毒(Ad)中的免疫印迹(Ad)和对照腺病毒(Ad)转化生长因子(TGF)-β表达的定量。MCT和ADBMPR2 + FAB-9B9治疗大鼠。将值计算为各种TGF-β信号与管家蛋白(α-微管蛋白)的化学发光的比率,然后表达与盐水组的比例的%变化。数据以平均值±表示SEM..*:P <0.05相对盐水;#: MCT组和Ad+Fab-9B9对照组p<0.05相对MCT和ADBMPR2 + FAB-9B9。在每个处理组中显示N = 6只动物的肺TGF-β和α-微管蛋白表达的代表性免疫印迹如图所示。用于B)盐水控制,C)MCT和对照AD + FAB-9B9和D)MCT和ADBMPR2 + FAB-9B9动物的TGF-β染色的小动脉染色的小动脉染色的高功率显微照片。比例尺=50μm以下。
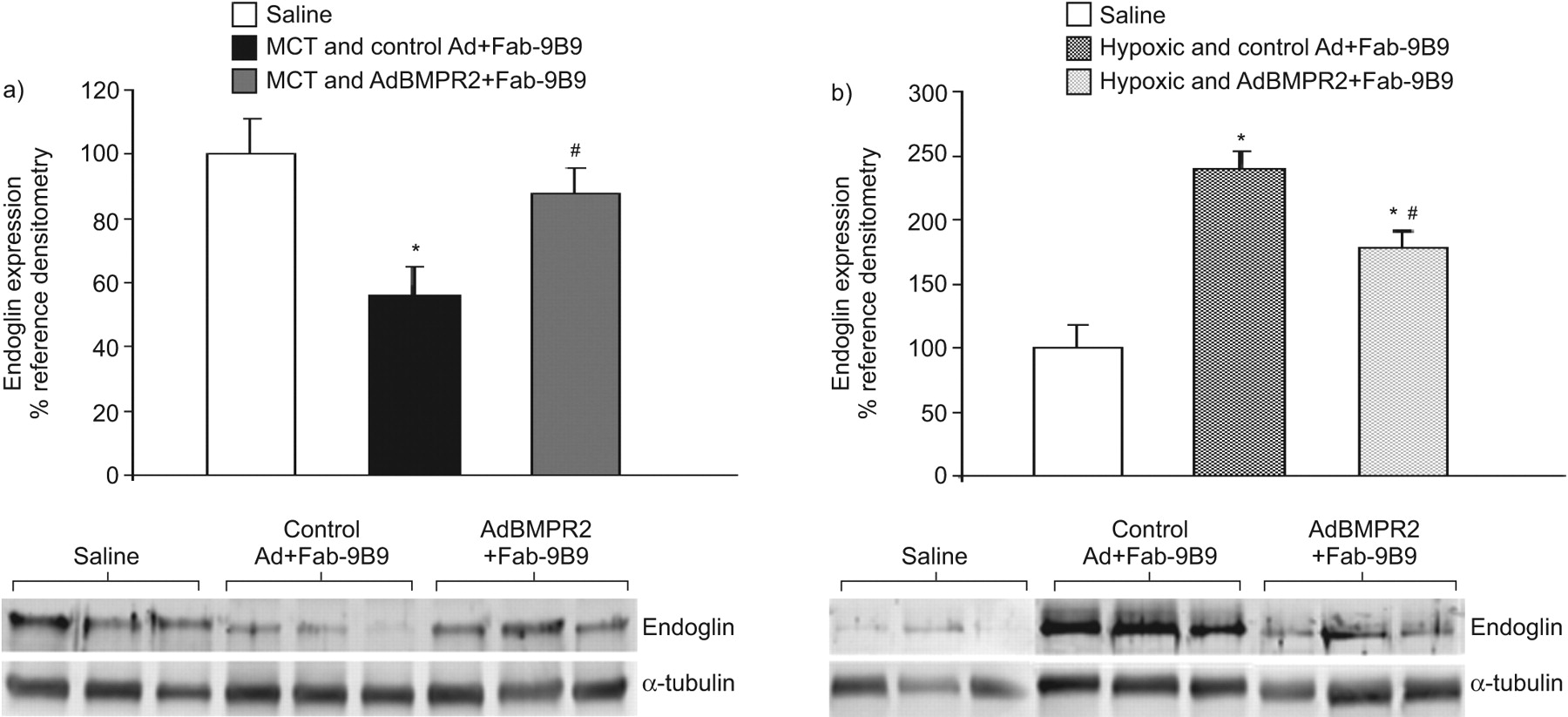
使用免疫印迹分析,我们无法检测TGF-β受体1或2的表达在任一模式的差异(数据未显示)。然而,内皮糖蛋白表达的分析表明MCT后显著减少(平均值±SEM.100.0±26.7%相对生理盐水组为55.9±20.1%相对MCT和控制AD + FAB-9B9分别;P <0.05)(无花果。6A).AdBMPR2治疗减轻了这种反应(87.7±19.7%;p < 0.05)。缺氧组内皮素表达显著升高(100.0±43.9%)相对盐水中239.5±35.5%相对缺氧组和对照组分别为Ad+Fab-9B9;P <0.05)(无花果。6B.).同样,AdBMPR2治疗减轻了缺氧模型中出现的升高(178.5±33.5%;p < 0.05)。
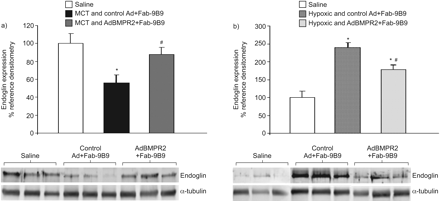
通过免疫印迹图像分析定量a)野百合碱(MCT)和b)缺氧处理的大鼠中内皮素的表达。BMPR2和α-微管蛋白表达的代表性免疫印迹分别如下图所示。计算值为相应内皮素信号相对于管家蛋白(α-微管蛋白)的化学发光比率,然后表示为与生理盐水组相比比例变化的百分比。数据以平均值±表示SEM..N = 6只动物每个治疗组中使用。*:P <0.05相对盐水;#:P <0.05处理和对照腺病毒(AD)预孵育与抗病毒/抗血管紧张素转换酶双特异性抗体缀合物(FAB-9B9)相对治疗和ADBMPR2 + FAB-9B9。
TGF-β1对HPMVEC细胞形态/ ENDMT的影响
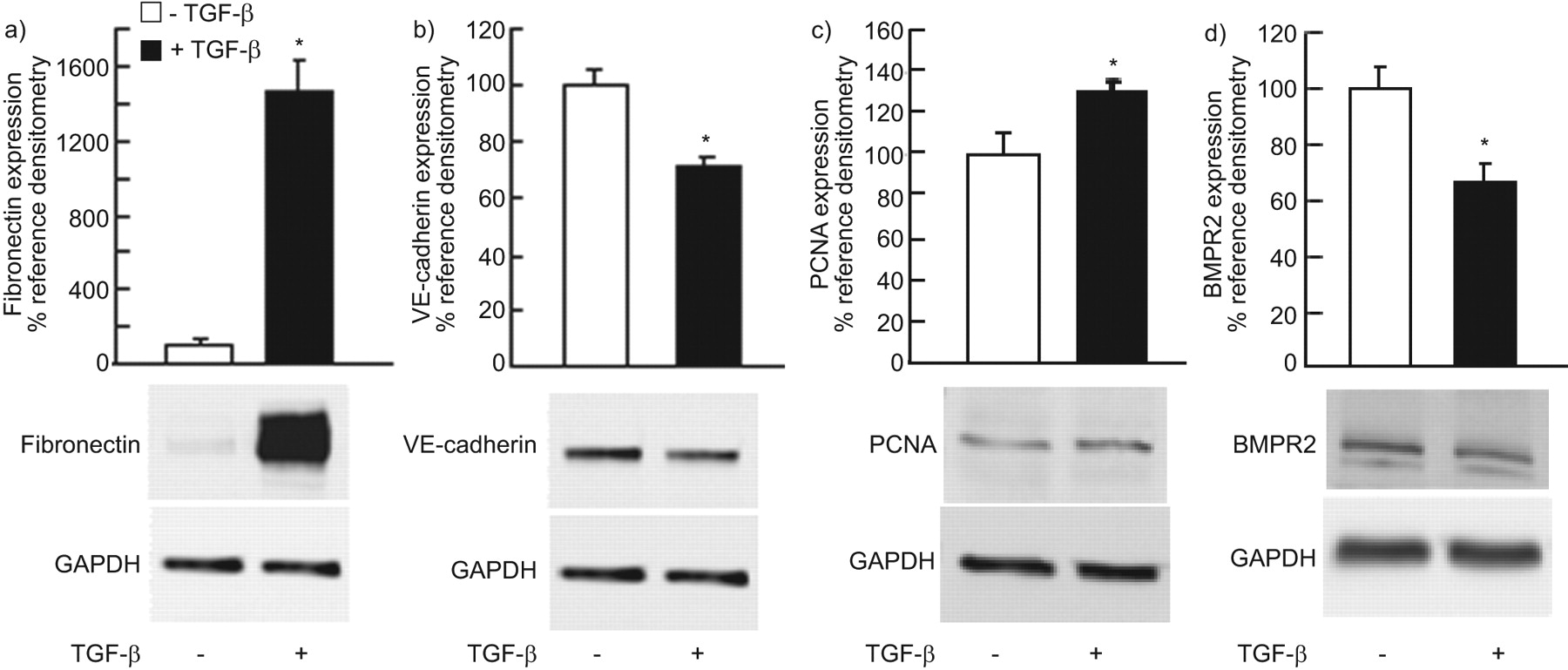
接下来,我们希望研究TGF-β对HPMVECs的生物学作用,以及在此背景下刺激BMPR2的影响。在对照组(不含TGF-β1)条件下,HPMVECs呈鹅卵石样外观,细胞间黏附明显(图7).TGF-β1 (5ng·mL)暴露于HPMVECs−1)显示细胞与细胞接触减少,细胞形态改变为细长的纺锤形成纤维细胞(图7 b).时间进程研究表明,TGF-β1治疗20天是诱导HPMVECs中EndMT的最佳治疗方案(数据未显示)。即TGF-β1处理诱导间充质标记物表达上调(例如纤连蛋白和S100A)与内皮细胞标记物的伴随下调VE-钙粘蛋白(图7 ck).在未处理的HPMVEC中检测到纤连蛋白和S100A的最小基础表达。在用TGF-β1处理后20天处理,HPMVEC纤连蛋白和S100A染色在纺锤形成纤维细胞型细胞中较强。检测到Ve-Cadherin的高基础表达,在TGF-β1处理细胞中变得显着降低。
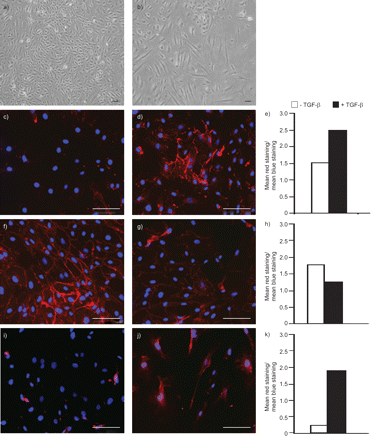
转化生长因子(TGF)-β1治疗对人肺微血管血管内皮细胞(HPMVEC)形态和蛋白质表达的影响。在a)缺失和b)在正常生长培养基中生长的Hpmvecs的相位对比显微照片,B)TGF-β1的存在(5 ng·mL−120天)。红色荧光表示C-E)纤连蛋白,F-H)血管内皮钙粘蛋白和I-K)S100A4的C,F,I)对照和D,G,J)TGF-β1处理的HPMVecs。4',使用6-二氨基-2-苯基吲哚(DAPI)染色来检测核(蓝色)。秤杆=100μm。e,h,k)使用图像分析软件定量正红色免疫荧光。为每个照片计算平均红色荧光强度,并通过将这种强度除以平均蓝色强度来归一化。
Endmt标记的免疫印迹分析
为了量化EndMT的标记物,我们使用TGF-β1 (5 ng·mL)处理20天的HPMVECs的总细胞裂解液−1每48小时补充一次,进行免疫印迹测定。TGF-β1治疗显着增加了间充质标志物表达;纤连蛋白表达增加14倍(图8)和Vimentin几乎加倍(n = 2;数据未显示),伴随的Ve-cadherin表达30%(无花果。8B.),与免疫荧光结果一致。用TGF-β1处理的HPMVEC的增殖反应增加了30%(无花果。8C.).
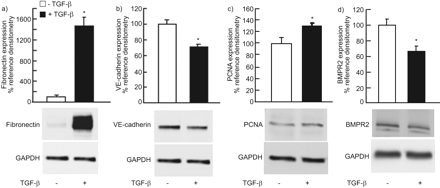
转化生长因子(TGF)-β1处理(5ng·mL−120天)增加了内皮 - 间充质转变的标记,a)纤连蛋白,内皮标记物和B)血管内皮(Ve)-cadherin。TGF-β1治疗还增加人肺部微血管内皮细胞(HPMVEC)C)增殖反应和降低D)骨形态发生蛋白受体II型(BMPR2)表达。值计算为内部蛋白质(甘油醛磷酸盐脱氢酶(GAPDH)的相应标记信号的化学发光比例,然后表达为来自未经处理的HPMVEC的处理HPMVEC的比例的百分比变化,IE。在正常生长培养基中生长的细胞。数据以平均值±表示SEM.三个独立的实验。免疫印迹来自一个有代表性的实验。PCNA增殖细胞核抗原。*:P <0.05相对对照组(无TGF-β1)。
TGF-β1对HPMVECS的BMPR2表达的影响
已经证实了TGF-β的增加和大鼠模型中BMPR2的减少(以及用BMPR2上调的TGF-β降低),我们接下来评估了TGF-β1对HPMVEC的影响。用TGF-β1孵育细胞导致平均值的减少SEM.BMPR2蛋白水平为65.8±10.9% (无花果。8D.).
BMPR2配体rhBMP-7和rhBMP-2逆转TGF-β1诱导的EndMT
为了确定在EndMT的BMP轴的上调的作用,我们在BMP-2和BMP-7的存在下重复对TGF-β1诱导EndMT研究。BMP-2和BMP-7的剂量为67%比TGF-β诱导的EndMT [的心脏纤维化的细胞培养模型中使用以下22.].同样,TGF-β1诱导纤维连接蛋白表达显著上调(160 - 200倍;无花果。9Ac)和Ve-cadherin表达减少(36-39%;无花果。9B.d).在TGF-β1的持续影响下,加入rhBMP-2 (333 ng·mL)−172 h)和rhBMP-7 (325 ng·mL−172小时)基本上抑制TGF-β1诱导的纤连蛋白产生;与单独的TGF-β1相比,纤连蛋白产生分别为33%和51%(无花果。9A和c)。血管细胞的完整性似乎分别由BMP-2和BMP-7完全或部分恢复,因为两种治疗显着逆转Ve-Cadherin表达的TGF-β1分别将59%和21%(无花果。9B.和d)。当将HPMVECS与单独的BMP-2或BMP-7一起孵育HPMVEC(数据未显示)时,检测到纤连蛋白或Ve-Cadherin的基础表达没有差异。
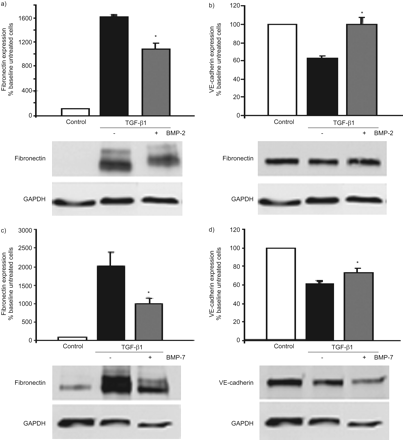
转化生长因子(TGF)的骨形态发生蛋白(BMP)-2,BMP-7的影响免疫印迹分析-β1诱导的人肺微血管血管内皮细胞(HPMVECs)内皮 - 间充质转换。用重组人(rh)BMP-2)治疗显著减弱由TGF-β1,b)中诱导纤连蛋白表达的增加恢复血管内皮(VE)-cadherin表达到基线值。的rhBMP-7处理,c)中基本上反转TGF-β1诱导纤连蛋白的生产和d)部分恢复内源性VE-钙粘蛋白的蛋白表达。值被计算作为纤连蛋白的化学发光的比率或VE-钙粘着蛋白在管家蛋白信号(磷酸甘油醛脱氢酶(GAPDH)),然后表示为与未处理HPMVECs处理HPMVECs的比率%的变化,IE。在正常生长培养基中生长的细胞。数据以平均值±表示SEM.三个独立的实验。免疫印迹来自一个有代表性的实验。*:P <0.05相对转化生长因子-β1治疗。
讨论
两者之间的因果关系BMPR2现在根据来自转基因小鼠模型的众多人类遗传研究和支持性证据来接受突变和PAH [23.,24.].这一发现的一个合乎逻辑的延伸是,BMPR2途径可能是治疗操纵一个新的目标,尽管这种策略很少受到关注至今。我们以前的工作确定了BMPR2基因传递靶向肺的内皮通过在一项预防性研究设计中,抗ace单克隆抗体(mAb)可改善缺氧诱导的肺动脉高压[14.].
在目前的研究中,我们从几个方面扩展了之前的发现。我们现在表明,BMPR2上调在两种互补的PAH动物模型中对建立PAH具有治疗作用。这些发现与之前的研究完全一致,即缺氧和MCT模型系统中的PAH都与BMPR2的下调有关[12.,13.].重要的是,现在有充足的证据表明,BMPR2在人类疾病的背景下在许多情况下下调BMPR2突变,从而扩大针对该途径的治疗的潜在适用性[6,25.- - - - - -27.].尽管近年来,基于前列环素类似物、内皮素受体拮抗剂和磷酸二酯酶5抑制剂的使用,PAH的管理取得了显著的改善,但仍有许多患者最终在这些治疗中失败,并正在寻找新的方法(例如,目前使用伊马替尼的临床试验)。综上所述,我们的发现为考虑基于BMPR2调节的治疗的发展提供了理论基础。
BMPR2功能障碍与肺血管疾病发展之间的机械联系仍不清楚。使用缺氧和MCT模型的最近的研究已经证明了BMPR2水平降低和异常的BMP / TGF信号传导,而最近,已经显示TGF-β在MCT模型中特别上调[17.,28.,29.].在这里,我们证实了在两个模型系统中肺BMPR2蛋白表达的减少,并表明这种减少是由BMPR2基因传递。我们发现在MCT模型中TGF-β显著增加,而在MCT模型中则相反BMPR2治疗。在缺氧模型中没有看到TGF-β的增加。其他人的TGF-β在这些模型中的作用的相对差异已被其他人注意到[17.].long.等等。[17.发现,在MCT模型中,TGF-β基本上增加,并且使用小分子阻止TGF-β受体(Activin-like激酶5)减少的PAH。相比之下,这种阻滞对缺氧诱导的PAH没有影响。在我们的研究中,减少BMPR2表达的校正在两个模型系统中都显示出益处。
目前研究的一个不足之处是,在涉及我们所看到的生理和重塑效应的确切细胞信号通路方面存在不确定性BMPR2基因传递。显着生理结果的证明提供了进一步询问这些机制的理由。在我们以前的工作中,我们调查了是否BMPR2转导用某种方式采取的方法来保护内皮细胞凋亡,因为它已经假设,疾病发病机会可能从增加的凋亡阶段开始,导致抑制抗性克隆的出现。这种早期凋亡的机制在人类PAH的发展中仍然是未经证实的,尽管有证据表明既定疾病中的相对抗凋亡[30.].出乎意料的是,我们以前在大鼠缺氧模型中发现和人类微血管细胞培养物转导BMPR2导致细胞凋亡增加。因此,我们推测,在已确定的疾病中,可能是BMPR2转导正在影响内皮通过内皮衍生的亲增殖因子的下调或对Endmt产生影响。
BMP信号包括配体启动的细胞表面BMPR2和BMPR1的异源二聚化(A或B),后者的激活,然后一系列受体调节的Smad蛋白的磷酸化(例如Smad1、Smad5和Smad8),进而与共同伴侣Smad (Smad4)复合物并转位到细胞核内以调节靶基因的转录[31.].在PAH开发的背景下,提出了TGF-β细胞信令似乎主要涉及SMAD2和SMAD3信号传导[32.],尽管在这个问题上的文献数据存在冲突,但与ZAkrzewicz.等等。[28.显示MCT模型中该通路活性降低,而Long.等等。[17.显示增加。造成这种明显差异的原因尚不确定,但可能是由于相对于疾病发作的测定时间的不同。UPTON等等。[32.]最近提出TGF-β-Smad2 / 3的信令和BMPR2-的Smad1之间的平衡/ 5/8信令可以是在控制肺脉管系统的细胞应答非常重要的。不幸的是,用福尔马林固定组织的分析技术限制在防止或Smad蛋白的p38-MAPK机制精确表征生理研究中使用的大鼠衍生的,这是正在进行的研究的主题。然而,我们确实发现在内皮因子表达水平稳健变化,在MCT缺氧模型明显的差异。我们在MCT模型降低内皮因子的发现与由Z报道mRNA的结果是一致的Akrzewicz.等等。[28.].内皮糖蛋白充当共受体的TGF-β信号传导和最近已经显示出增强的Smad1的TGF-β诱导的磷酸化/ 5/8 [33.].我们发现BMPR2的上调与MCT模型中内源性蛋白表达的恢复有关。据我们所知,在大鼠缺氧模型中,虽然缺氧已被证明会增加内皮素在人内皮细胞中的表达,但内皮素水平以前没有报道过体外,在心肌和胎盘缺血的模型中[34.].因此,内脏在两只大鼠PAH模型中的作用似乎是完全不同的,BMPR2和内阴光引出之间的链接需要进一步调查。
BMP/BMPR信号转导系统是控制细胞分化和凋亡的关键。前者使用Smad信号通路,而后者通过激活p38-MAPK发挥作用。现在也知道,这些受体形成预先形成的受体复合物,配体与之结合,导致Smad信号,或BMP配体启动受体复合物,导致p38-MAPK的激活[35.].发生预先形成的或配体引发的复杂形成是否取决于细胞表面上的BMPR2和BMPR1A分子的相对数量[36.].此外,已经有人提出,当BMPR2表达水平低于正常表达的10%的临界值时,异常信号就会盛行[32.].这一现象在缺氧和MCT模型中均有报道[13.,37.],其中BMPR2表达已经下降,导致正常的SMAD1 / 5/8信令几乎废除。在我们的研究中,我们在BMPR2与PAH发展相关联的明显下降,尽管〜25%的幅度小于先前报道的程度。这些差异可能是由于使用的技术的分析或差异的问题。在这里,我们已经分析了组织切片中的总表达,这可能会低估内皮细胞的相对幅度,但分析细胞水平的BMPR2表达需要更多的工作。广告矢量BMPR2在我们的研究中,靶向肺内皮细胞的释放恢复了BMPR2的整体水平,因此,可能是细胞野生型受体可用性的平衡。
基因递送的成功可能依赖于转导的特定靶细胞,特别是当递送膜结合受体的基因时。BMPR2在平滑肌细胞上的表达是重要的,许多研究表明了在环境中表现出存在的功能障碍平滑肌增殖BMPR2突变。我们针对内皮细胞的方法并没有直接解决这个问题。然而,与我们的工作相反,体内肺血管平滑肌的转导通过在MCT模型中测试时,气溶胶途径不会导致任何生理改进[38.,这可能支持内皮细胞在这种情况下表达的重要作用。我们之前使用该基因传递技术和报告基因的工作并没有显示出明显的心脏表达,在目前的研究中我们无法通过免疫荧光检测到转基因在心脏组织中的表达。因此,我们认为积极的结果是由于对肺血管的影响,而不是任何直接的心脏影响。如果一种基因传递策略要实现临床应用,还需要更多的工作来确定最佳的方法。
最近,TGF-β诱导的EndMT作为纤维化性肺疾病的机制得到了推动[39.].还研究了Endmt在血管疾病中潜在的作用。响应于TGF-β1的那些表明Endmt的研究已经使用来自较大血管的动物原代内皮培养物[40,41.或人类真皮内皮细胞[42.].基于其他人报告的给药和时间课程研究[22.,43.,我们选择了5 ng·mL的给药方案−1在正常生长培养基和18-21天的接触期间测试TGF-β1是否可以在更相关的HPMVEC中诱发ENDMT,以及此报告,随后用TGF-β1刺激,HPMVECS展示了他们鹅卵石形态的变化来主轴 -形状,成纤维细胞样形态。我们还观察到内皮标记表达(Ve-Cadherin)的丧失,其增益表达间充质标记物(纤连蛋白和S100A4)。特别重要的与目前的研究,我们发现TGF-β1刺激导致BMPR2表达的减少。虽然我们已经评估了我们的BMPR2在这种情况下,这些细胞(在培养中不表达ACE)的转导效率低,阻碍了研究。因此,我们通过添加外源性bmp来恢复BMPR2/TGF-β信号通路的平衡,并为bmp在TGF-β诱导的EndMT中的保护作用提供了证据。我们的研究结果表明,即使在TGF-β1持续存在的情况下,BMP-2和BMP-7也会导致EndMT的逆转,这与心肌纤维化模型一致,其中rhBMP-7传递抑制EndMT和心肌纤维化的进展[22.].我们用来显示效果的bmp剂量是心脏研究中认为的最佳剂量的三分之一,但我们没有看到低于这个剂量的显著效果。目前,EndMT在PAH中的作用仍然是一个新假设,在该领域引起了重大的兴趣,正如最近的综述所指出的[44.- - - - - -46.].缺氧诱导的肺血管重塑中显示了肺动脉源性内皮细胞转化为平滑肌状细胞的转化性[41.].在其他情况下也发现了对EndMT概念的支持,包括心肌纤维化、动脉粥样硬化、伤口愈合和血管发育[44.].然而,EndMT在人类PAH中的作用还没有确定的证据。
发展的主要障碍BMPR2基因治疗当然是将我们的方法转化为一种在人类临床环境中可行的策略。人类疾病的基因治疗方法通常难以产生显著的临床影响。这种困难很大程度上与用于基因传递的载体技术的局限性有关。在目前的研究中,我们使用我们之前建立的方法靶向肺内皮细胞通过抗ace单克隆抗体,我们已经证明它可以改善肺血管的基因传递[20.,21.].我们的方法与载体的气溶胶递送相比取得了有用的结果,这在MCT模型中没有减少PAH [38.].重要的是,我们的策略实现了内皮细胞转导(肺中BMPR2的主要表达位点),而Mc米urtry等等。[38.]旨在血管平滑肌。然而,我们目前的工作实质上仍然在原则上阶段。增强BMPR2表达所需的持续时间来实现治疗结果BMPR2突变还不清楚。BMPR2突变的渗透性渗透有限,用于引起临床疾病(<20%的突变载体发育疾病),通常接受某些形式的“第二次命中”是必要的。也许瞬态BMPR2过表达,即使是短期数周,也可以中断病理级联。我们这里使用的第一代AD载体只能达到短期表达(通常在免疫活性动物中短数周数),并且由于中和抗体的发育而不适合重新管理。然而,随着载体技术的进一步改进,可以利用当前的载体技术钻点来实现显着的生理结果的事实。传染媒介技术的演变在经转基因次备体载体,重组靶向适配器的发展以及病毒的修饰中已经很好地进行,以减少肝脏摄取和规避预先存在的免疫力[47.,48.].较长期的转基因表达可能与辅助依赖性Ad载体,向其中可应用于靶向技术[来实现49.].在使用细胞的情况下,随着用内皮酰化氧化氮合成酶转导的基因递送载体和细胞的兴趣也在I临床试验中[50.].或者,可以针对该途径引用更多常规的药物。例如,一些BMPR2突变导致产生由于异常折叠而无法到达细胞膜的潜在官能蛋白质。改进的贩运,药理学伴侣,如尾剂或甘油,其具有重定向BMPR2突变体的疗效,可以帮助恢复患者子集中的BMPR2信号传导[51.].最近国家登记研究得出的相当令人失望的存活率数字清楚地表明,需要不断发展新的治疗方法,以补充现有的战略。
总而言之,目前的研究提供了证据,表明使用基因传递方法增加BMPR2表达可以改善两种常见大鼠模型的生理结果。还需要更多的研究来确定这种方法是否与人类疾病有关。
脚注
有关编辑评论,请参阅325..
这篇文章有提供补充材料www.www.qdcxjkg.com.
支持声明
这项研究是由国家健康和医学研究委员会项目拨款和从业者奖学金,以及澳大利亚国家心脏基金会资助的。
感兴趣的语句
对S.M. Danilov感兴趣的声明和研究本身可以在www.www.qdcxjkg.com/site/misc/statements.xhtml
- 收到了2010年12月5日。
- 接受2011年6月24日。
- ©2012年











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}