





摘要
慢性阻塞性肺疾病(COPD)通常被认为依赖于对有毒或传染性病原体的异常免疫反应,这可能导致慢性炎症。然而,这种异常反应的起始机制尚不完全清楚。在这里,我们为COPD的开始提出了一个新的假设,即对吸入制剂(主要是香烟烟雾)的免疫反应是针对气道上皮,由于肺上皮屏障细胞(LEBCs)的氧化DNA损伤。该模型的步骤如下。1)卷烟诱导lebc的DNA氧化损伤。2)获得性突变在lebc的微卫星DNA水平上表达。3)改变的lebc被树突状细胞(DCs)识别为“非自身”。DCs带着新的信息,移动到淋巴结,将其呈现给naïve t淋巴细胞。4)出现主要的CD8+细胞毒性t淋巴细胞增殖。CD8+细胞通过释放穿孔素和颗粒酶攻击改变的lebc激活细胞死亡级联反应。
显然,上述情景还需要进一步的实验探索。然而,它是COPD异常炎症启动的一个有吸引力的模型,包括lebc的氧化DNA损伤和宿主免疫反应。
慢性阻塞性肺疾病(COPD)是世界性的主要健康问题,其发病率、发病率和死亡率都在不断增加1- - - - - -3..最近的指南强调,慢性阻塞性肺病的特征是进行性的,不是完全可逆的气流限制,这是肺部对吸入有毒气体和颗粒的异常炎症反应的结果2,3..绝大多数COPD患者共享慢性卷烟烟雾暴露,作为常见和主要的环境的疾病1- - - - - -5.香烟烟雾是颗粒、自由基和活性化学物质的主要来源,所有这些都会对肺部产生压倒性的氧化负担1- - - - - -3.,6.这些高反应分子的分子效应可以解释在COPD患者中观察到的一些病理:炎症、肺破坏和肺气肿、气道纤维化、粘液分泌过多和骨骼肌萎缩5.然而,只有少数吸烟者被诊断为临床相关的COPD1- - - - - -4,7,这表明慢性阻塞性肺病是宿主-环境相互作用的结果,很可能是遗传决定的8.在COPD中研究的候选基因中是影响蛋白酶和脱发酶的产生的基因,该基因调节烟雾的有毒物质的代谢,参与蛋白质清除和影响炎症介质的基因的那些8- - - - - -10.然而,似乎最不太可能是单一分子途径将占COPD的发病机制。
最近,表观遗传机制,如获得性体细胞突变可能是COPD分子发病机制的另一个基本因素11.体细胞突变不影响生殖系;它们不是可遗传的,尽管获得这种突变的易感性可能是由遗传基因控制的11.在人的一生中,基因组完整性受到各种类型的获得性DNA损伤,由外源性(例如紫外线、电离辐射、有毒化学物质和香烟烟雾)或内源性物质(例如正常代谢的副产品,如活性氧(ROS)和自由基、停滞的复制叉、减数分裂重组和免疫系统成熟)12,13.这些临界DNA病变可导致细胞死亡或各种遗传改变,包括缺失,易位,杂合性丧失和微卫星DNA不稳定性12,13.在正常情况下,细胞配备了许多修复途径,可去除损坏并恢复完整的DNA13.然而,增加和持续持续的氧化应激可以使人DNA错配修复(MMR)系统失活,导致获得突变。这些突变永久性地改变了DNA嗜睡能力,导致基因组不稳定性14.吸烟引起的体细胞突变一旦获得,将持续数年或数十年15.同意以上假设,在COPD患者的微卫星DNA中检测了细胞遗传改变16- - - - - -18.
在本综述中,我们提出了一种新的COPD启动假说,即与吸烟相关的氧化应激可能损伤肺上皮屏障细胞(lebc)的DNA,进而被宿主免疫系统误用为“非自我”。一种非典型的适应性免疫反应产生,CD8+细胞毒性细胞占优势,导致细胞死亡级联反应(图1)⇓)。
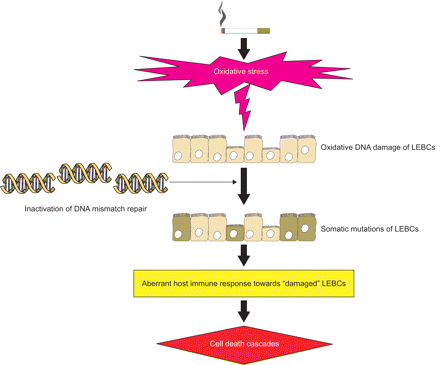
慢性阻塞性肺病开始的提出模型。吸烟的氧化负担会破坏肺上皮阻隔细胞(LEBC)的DNA,并灭活人DNA错配修复系统,导致Lebcs的躯体突变。改变的Lebcs被宿主被认为是“非金属”,诱导对改变的Lebcs的异常免疫应答,导致细胞死亡级联的激活。
假设
吸烟、氧化应激、肺部炎症与慢性阻塞性肺病之间的联系已经被广泛研究,并提出了多种理论来解释慢性阻塞性肺病的发病机制19- - - - - -26.在本文中,我们提出了一种新的COPD发病假说(图1)⇑)。
具体来说,在慢性阻塞性肺病发病初期,吸烟会影响空气-肺屏障系统,特别是其细胞成分通过反复氧化应激导致氧化DNA损伤27- - - - - -30..氧化应激损伤lebc的DNA,导致获得性体细胞突变11,14,15,进一步抑制DNA MMR系统的关键基因,影响DNA的自修复能力11,14.
lebc的体细胞突变是通过改变的细胞表达的危险/警报信号来激活和极化树突细胞(DC)网络的一种间接方式(图2)⇓)31,32.在“危险模型”中,树突状细胞(如树突状细胞)暴露于病原体、毒素、机械损伤和香烟烟雾后,可被自残细胞产生的危险/警报信号激活。这个模型得到了内生、非外来报警信号的发现的支持32,包括改变的哺乳动物DNA,RNA,热休克蛋白,干扰素(IFN)-A,白细胞介素-1B,透明质的CD40-L和分解产物32,33..
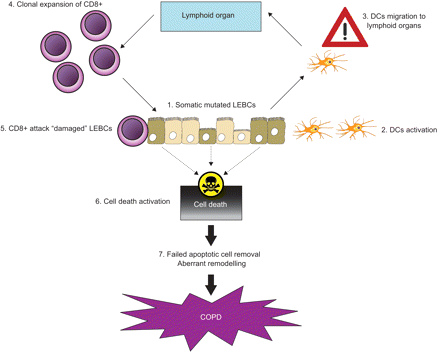
慢性阻塞性肺疾病(COPD)中肺上皮屏障细胞(lebc)获得性体细胞突变1)体细胞突变的lebc被宿主识别为非自身。2)树突状细胞(DCs)被改变的lebc发出的“危险/警报”信号激活,然后3)树突状细胞前往引流淋巴结呈现新的信号。4) CD8+细胞毒性t淋巴细胞增殖,然后5)CD8+ t细胞迁移到最初受损伤的部位,通过释放穿孔素和颗粒酶攻击改变的lebc, 6)激活细胞死亡。7)凋亡细胞清除失败可能会阻碍炎症的解决和肺泡完整性的维持,从而导致COPD发病。
随后,DCs带着受损lebc产生的内源性警报信号到达引流淋巴结,并将其呈现给naïve t淋巴细胞,诱导CD8+细胞毒性t淋巴细胞增殖34.- - - - - -36..CD8+ t细胞迁移到最初损伤的位点,并通过释放穿孔素和颗粒酶攻击改变的lebc。穿孔素在靶细胞的细胞膜上形成孔,而颗粒酶,如丝氨酸蛋白酶,进入靶细胞的细胞质,改变它们的功能和/或激活细胞死亡(图2)⇑)37.- - - - - -39..
支持这一假设的研究
COPD中lebc的DNA氧化损伤
对呼吸道有毒或传染性病原体的防御是由许多不同的机制提供的。在气道中,lebc构成第一道防线,在与吸烟有关的疾病中,lebc的数量和功能有所不同40,41.,如copd1- - - - - -3..吸烟者用每支香烟暴露于数千种激进的和反应化学品,因此认为氧化应激是COPD发病机制中的中央组分5.ROS、活性氮(RNS)和碳中心自由基是烟雾焦油和气相中的组成成分5.此外,还存在ROS的内源性来源,例如由活化的肺泡巨噬细胞和中性粒细胞产生的那些响应烟雾,这进一步促进上皮细胞损伤5.增加ROS生产与蛋白质,DNA和脂质的氧化直接相关,这可能导致直接肺损伤或诱导各种细胞反应,通过产生次级代谢反应性物种和ROS介导的分子机制12- - - - - -15,27.ROS可以攻击核酸,导致碱性修饰,双基损伤,链断裂,错配和交联,导致体细胞突变水平增加11,14,27.在大多数情况下,这些突变可以通过内源性修复过程来纠正,例如人类DNA MMR系统。然而,由自由基引起的翻译后修复酶的失活可能会增强DNA损伤的积累14.有趣的是,女性吸烟者对卷烟烟雾的新陈代谢加速了导致DNA加合物和突变的负担增加,而是降低了DNA修复能力42.,43.因此,他们更容易受到体细胞突变的影响,这可能导致慢性阻塞性肺病的发生42.,43..
最近的研究报道了COPD患者外周血中氧化应激和DNA损伤水平的增加29,30..此外,在痰细胞中也有微卫星DNA不稳定性的报道16- - - - - -18和组织15,44.来自COPD患者和吸烟者,可能反映了DNA MMR系统的缺陷14.
lebc获得性体细胞突变
目前和以前吸烟者肺上皮的分子损伤已经被证实,并且似乎在戒烟后仍然存在11,15.在COPD患者的痰细胞中发现微卫星DNA不稳定16- - - - - -18,45..尽管痰液是一个相对异质性的样本,但研究表明微卫星不稳定性只存在于痰液上皮细胞亚群中45.,46.,由免疫磁分离隔离并进一步加工用于基因组分析46..
微卫星改变涉及多态微卫星DNA序列的简单核苷酸重复大小的改变,导致一个或两个等位基因的电泳迁移性的改变47..这些DNA序列的插入或删除与高体细胞突变率相关,并与缺陷的DNA MMR系统有关48..MMR系统是已知的DNA修复机制,非常有效地在鉴定和随后在DNA复制中纠正误差,以在复制过程中逃避DNA聚合酶校对49..MMR校正基质替换和帧误差,以及微卫星序列中的扩展和收缩。特别地,MMR系统积极参与内源性产生的DNA病变和DNA损伤信号响应的修复49..研究表明,与吸烟和慢性炎症相关的氧化应激可能破坏MMR系统的蛋白质成分,导致其功能失活14,48.,50.,51..常等等。14报道了非胞间毒性水平2O2以剂量依赖的方式使MMR系统的单碱基错配和环修复活性失活,而这种失活最可能是由于hMutSa、hMutSβ和hMutLα蛋白复合物的氧化损伤。在一些模型系统中,氧化性DNA损伤可导致微卫星滑移突变50.,51..据报道,在人类细胞系中,DNA氧化损伤后,微卫星突变的增加高达27倍51..此外,Makris等等。18显示了微卫星DNA不稳定性和慢性阻塞性肺病加重之间的显著关联,表明DNA MMR系统改变和慢性阻塞性肺病频繁加重或氧化DNA损伤之间的联系反之亦然18.
对改变的Lebcs的宿主免疫应答
各种形式的细胞损伤导致DNA碎片和破坏修复潜力,使其成为“外国”52.,53..该假说认为,一旦lebc遭受氧化性DNA损伤,它们就会被免疫系统的主要传感器DCs检测为“非自我”54..DCS是肺部的主要抗原呈递细胞,填充整个呼吸道树,可以细分为两种类型:髓样DC和血浆骨质DCS(PDC)54..
在危险信号存在或不存在的情况下,在识别自身或非自身信号的基础上,DC抗原摄取和呈递的相互作用导致免疫沉默或免疫激活特性,从而指示肺内耐受或防御免疫的结果54..
在外周中的抗原取样后,DC在CC趋化因子受体(CCR)7依赖性时尚中迁移到区域淋巴结,在那里它们激活NaïveT细胞以增殖55..树突状细胞和自然杀伤细胞(NK)似乎相互调节对方的成熟56..NK细胞与PDCs或髓系树突状细胞的相互作用深刻地影响了先天抵抗功能56..在肺稳态条件下,PDC和骨髓DCs之间存在平衡,使抗原耐受性并避免明显炎症56..在适应性免疫中,树突状细胞从循环中招募,并向上皮表面迁移,在那里它们捕获抗原并识别“危险信号”。随后,它们迁移到区域引流淋巴结,并将隔离的抗原提供给naïve淋巴细胞,以诱导初级免疫应答或耐受,这取决于共刺激分子表达、细胞因子释放和它们的成熟状态57.- - - - - -59..
我们认为树突状细胞极有可能参与慢性阻塞性肺病的发病机制,不仅在慢性气道炎症的起始阶段,而且在其特有的慢性气道炎症模式的延续过程中55.,60.,61..一旦被激活,DCs通过化学引诱剂(包括作用于细胞表面受体(如CCR7)的巨噬细胞炎症反应蛋白(MIP)-3β)促使其成熟并迁移到引流淋巴结55..根据初始刺激的性质,DCs将与naïve淋巴细胞合作,诱导三种主要反应之一:辅助t细胞(Th) 1型、Th2型或t调节型62..
诱导主要是CD8+细胞毒性I型t淋巴细胞增殖
研究表明,在COPD中,DCs有助于诱导主要是CD8+细胞毒性I型t淋巴细胞应答23,34.,54.,63..转变为主要的I型免疫反应诱导慢性气道炎症,CD8+细胞数量增加,释放IFN诱导蛋白10、IFN-γ和肿瘤坏死因子(TNF)-α,以及穿孔素和颗粒酶22,34.,35..此外,b细胞和CD8+ t细胞的分布范围和体积都在增加,并组织成淋巴滤泡22,24,64..在吸烟后发育肺炎症和进步肺泡空气扩大的小鼠中检测到类似的淋巴卵泡65..
自我抗原的交叉呈递(例如损伤的lebc)由DCs到t细胞可能导致自身免疫的发展57.- - - - - -59.,66..此外,在DC成熟中的香烟吸烟诱导损伤可增强CD8 + T细胞增殖通过外来和/或自身抗原交叉表达增加67..此外,DC成熟降低可能使肺部CD4+/CD8+ t细胞比例向CD8+细胞倾斜,因为DC启动CD8+增殖程序所需的时间小于CD4+细胞所需的时间68..由于CD8+细胞的增殖能力大于CD4+细胞,CD4+细胞更容易受到DC成熟降低的影响69..CD4+ t细胞促进CD8+ t细胞向记忆细胞的分化。吸烟严重程度、气道阻塞程度、肺气肿均与CD8+细胞和/或CD8+/CD4+比值升高有关67.- - - - - -69..
在相似之处对病毒感染的免疫反应,CD8 +细胞攻击改变的Lebcs并进行细胞毒性功能,激活细胞死亡级联20.,26,37..对此,Chrysofakis表示同意等等。37.显示来自COPD患者的CD8 + T细胞产生更多的穿孔素,是极其细胞毒性的。最近,Vernooy.等等。39.研究GrA和GrB在COPD患者肺组织中的蛋白和mRNA表达,在CD8+ t细胞、CD57+ NK细胞、II型肺细胞和肺泡巨噬细胞中发现GrA和GrB。GrA和GrB都参与了穿孔素依赖过程,导致DNA片段和穿孔素依赖事件的启动,导致细胞凋亡39..凋亡,虽然性质中的抗炎性,如果凋亡体未及时除去凋亡,可能会引发自身免疫过程,可能导致持续炎症70.
然而,我们应该指出,大多数常驻细胞(lebc、巨噬细胞、内皮细胞和细胞外基质成分)在受损时,都能够诱导免疫反应和/或参与氧化负担71..
总结
吸烟和气道炎症可能导致氧化DNA损伤导致lebc的体细胞突变。这可能主要影响DNA MMR系统,导致lebc无法保持基因组完整性。受损的lebc被宿主免疫系统误用为“非自身”,导致细胞毒性CD8+细胞的克隆性扩增,导致细胞凋亡和/或坏死。然而,我们确实承认,纵向研究可以回答以下问题:易感吸烟者氧化炎症负担的增加是否与lebc体细胞突变和宿主免疫系统之间的联系。
感兴趣的语句
没有宣布。
- 收到了2008年4月30日。
- 接受2009年3月5日。
- ©ERS期刊有限公司











{kind=link}
{kind=link}
{kind=link}
{kind=link}