





摘要
儿童肺动脉高压(PH)种系突变的发生率缺乏文献记载。本研究的目的是确定儿科队列中PH基因的突变频率,并描述突变携带者的临床特征。
该研究涉及66例PH指数病例:35例特发性肺动脉高压(IPAH)、5例家族性PAH(FPAH)、3例肺静脉阻塞性疾病(PVOD)和23例先天性心脏病(APAH-CHD)合并PAH的儿童。
在23例APAH-CHD患儿中未发现突变。在40例IPAH或FPAH患儿中,发现了12个突变:5个在BMPR2;四点ACVRL1;还有三个TBX4。在三例PVOD病例中,两例携带EIF2AK4突变。突变携带者在诊断时病情更严重,需要更积极的一线治疗。这三名患有PVOD的患者在诊断时病情非常严重,需要进行肺移植。
儿科多环芳烃的遗传结构丰富ACVRL1和TBX4但需要进一步的研究来证实这些结果。儿童期发病的PAH在检测的某一基因突变的儿童在诊断时表现更严重,但结果与在非携带者中观察到的相似。
摘要
儿童肺动脉高压具有特定的遗传结构http://ow.ly/54yP301hCQi
介绍
肺动脉高压(PAH)是一种罕见的毁灭性疾病,其原因是血管细胞增殖和内膜纤维化导致小口径肺动脉逐渐闭塞,并导致心力衰竭,未经治疗的平均生存率<3% 成年和<10岁 儿童数月[1.–3.].PAH可以是良好识别的病理条件的并发症,它可以在家庭历史或遗传突变(遗传性PAH; HPAH)的背景下发生,或者在没有鉴定的预感因素的情况下被认为是特发性(IPAH)[4.].IPAH来源不明,约占成人多环芳烃的70%[5.].在前瞻性TOPP(小儿PH跟踪结果和实践)登记中,收集了儿科和青少年肺动脉高压的数据,88%的儿童患有PAH,其中57%的特征为IPAH或HPAH, 36%为PAH与先天性心脏病(APAH-CHD)相关[6.]HPAH是一种遗传异质性疾病。与成人HPAH相关的主要基因为BMPR2这是一种编码骨形态发生蛋白信号通路的2型受体的基因。低外显率占主导地位BMPR2在>中发现了70%的家族性PAH (FPAH)和15%的IPAH [7.,8.].这些突变的渗透在两种性别中不同(男性14%)相对根据L阿尔金等等。[9])。同一途径的其他基因发生突变,ACVRL1在极少数情况下eng,在大多数情况下可导致PAH与遗传性出血性毛细血管扩张症(HHT)相关[10,11].很少有突变SMAD基因家族,以及KCNK3和CAV1基因已被报道[12–14].最近,TBX4突变(小髌骨综合征)已被确定与儿童期发病的PAH相关[15].双腿隐性突变EIF2AK4在遗传性肺静脉阻塞性疾病(PVOD)和肺毛细血管血管瘤病的起源处发现了该基因,这两种疾病分别表现为以静脉病变或毛细血管增生为主的密切相关表型[16,17].
一些研究专门用于确定儿科PAH的基因突变arrison等等。[18)确定了两个BMPR2突出突变从头和两个HHT引起的基因突变eng和ACVRL1在16例IPAH患儿中Rosenzweig等等。[19)发现8BMPR278例PAH患儿中突变携带者(10%),其中大部分为fpa7 /8。在29例FPAH和IPAH患儿中,P法尔等等。[20.)发现BMPR2四个儿童突变(14%),两个突变ACVRL1,以及一种意义未知的变异(VUS)eng.FUjiwara.等等。[21]在16岁以下使用PAH进行了一系列21个证据,并发现了四种突变BMPR2(19%)和5个突变ACVRL1.识别PAH基因突变对于患有PAH的成年患者具有预后意义BMPR2或ACVRL1在这两组突变携带者中,突变在诊断时更年轻,到死亡的时间更短相对非承运人(10].儿科形式的多环芳烃,CHida.等等。[22]表现出相似的发病年龄和较差的预后局限于BMPR2死亡时间明显缩短的携带者,但这种差异在携带者中并不显著ACVRL1突变。
在APAH-CHD中,在大多数情况下,左右分流器与PAH之间的因果关系被认为是显而易见的。最后的国际分类定义了3型APAH-CHD,作为PAH,巧合CHD,包括小分流缺陷,不会引起严重的PAH,并遵循类似于IPAH的课程[23]PAH基因突变在这些患者中的作用尚不清楚,在任何类型冠心病修复术后发生PAH(4型)的儿童中也不清楚,这些儿童会发展为意外的肺血管疾病。对于患有APAH-CHD的儿科患者,遗传数据有限。P法尔等等。[20.]描述一eng2型APAH-CHD(左向右分流)患者不太可能出现致病性VUSBMPR2家族性PAH合并冠心病的突变病例。Harrison等等。[18]在两例APAH-CHD患者中未发现任何突变财大等等。[24描述三个BMPR23例2型APAH-CHD患儿的VUS。
考虑到最近发现的基因,关于儿童期发病PAH的遗传结构的数据有限。本研究旨在确定FPAH、IPAH、PVOD患儿和3型、4型APAH-CHD中肺动脉高压(PH)基因突变的比例,并描述其诊断时的基线临床特征和各自的预后。
方法
研究人群
2007年1月至2013年12月,纳入指标儿科患者(n=66);66例无亲属关系患者,年龄6.8±5.13岁(平均±sd)在他们的父母对本研究给予书面知情同意后,他们被纳入了PH诊断。所有患者均在同一儿科心脏科接受研究。PH诊断在右心导管插入术期间确定,定义为平均肺动脉压>25 静止时毫米汞柱,肺血管阻力指数(PVRI)>3个木单位·m2.平均肺毛细血管楔压>12 毫米汞柱。对所有儿童进行全面的诊断性检查,以检测可能易患PAH的共病。在所有病例及其父母中搜索HHT症状。患者按照2013年在法国尼斯商定的国际分类进行分类[4.].在临床和生物学调查排除所有已知原因后,PAH被认为是特发性的。当家族中至少有两例病例报告延伸至三级亲属时,这些病例被认为是FPAH。1型(艾森曼格综合征)和2型(左向右分流)APAH-CHD患者[23]由于血流动力学状态被认为足以解释多环芳烃的发生,因此未纳入。包括3型(n=13)和4型(n=10)APAH-CHD患者。排除畸形综合征或先天性代谢错误、已知非整倍体(21三体)和其他细胞遗传学异常(在染色体1和14上)(n=18)。患者在诊断时进行了超声心动图、N末端脑钠肽原(NT-proBNP)和循环内皮细胞测量(表1和2.) [25].发生PAH的病例是在纳入时诊断时进行基因分型的(33/40),而流行病例(7/40)在纳入时仍存活,但基因分型是在可变延迟(7±4.5年;均值±sd).同样,APAH-CHD患者的发病率(11/23)和患病率(12/23)。
对于18岁以下的儿童,父母批准后的症状个人(父母拒绝案件)提出了基因检测。当鉴定突变时,遗传测试在父母中进行以区分遗传从头突变。建议所有亲属进行基因检测和详细临床检查,以进行PH诊断。未受影响的兄弟姐妹在父母批准后,根据法国法律对未受影响的儿童进行基因分型。
我们的机构审查委员会和道德委员会批准了该方案。
基因分析
所有患者都进行了筛选BMPR2和ACVRL1点突变和大的重排。IPAH或FPAH患者,且无突变BMPR2或ACVRL1,进行进一步筛选EIF2AK4, TBX4和KCNK3点突变。eng和SMAD9对其他基因中未发现突变的FPAH患者进行筛查(图1).自从TBX4,KCNK3和EIF2AK4在研究开始时,我们不知道患者是否与PH有关,包括患者,我们对这些额外的基因进行了回顾性测试。
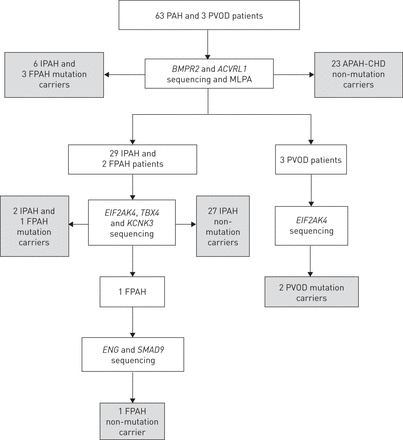
遗传分析方案流程图。每一个被检测的基因都与用于检测突变或重排的多重连接依赖探针扩增(MLPA®)的技术一起显示出来。每个基因携带突变的受试者的数量都显示出来。肺动脉高压;肺静脉闭塞性疾病;IPAH:特发性多环芳烃;FPAH:家族多环芳烃;PAH- chd: PAH与先天性心脏病相关。
点突变和大重排的筛选按Sztrymf等等。[7.].用于用于的底漆BMPR2,ACVRL1,eng,EIF2AK4,SMAD9和KCNK3已被描述[7.,10–12,14,16].用于用于的底漆TBX4筛选在补充表S1中提供。
核苷酸的编号遵循人类基因组变异协会的建议。所有确定的突变都用Alamut软件2.2版(交互式生物软件,法国鲁昂)进行了分析。
结果
所有患者每年至少接受两次标准化治疗,包括世界卫生组织功能分级、NT-proBNP和超声心动图测量(表2).考虑到以下事件的发生,以分析或组合:死亡;肺或心肺移植;Potts分流。
统计数字
数据以平均值±表示sd。XLSTAT 2014软件(美国纽约州纽约市Addinsoft)用于执行非参数Mann–Whitney U检验,比较有无突变的患者,或在适当情况下计算卡方检验;p<0.05被认为具有统计学意义。MedCalc统计软件版本12.7.7(MedCalc软件bvba,比利时奥斯坦德)用于构建Kaplan–Meier生存曲线(图2),并计算对数秩检验,该检验用于检验三种主要事件(死亡、移植、Potts分流)突变携带者和非携带者(仅事件案例)的自由概率分布的差异。
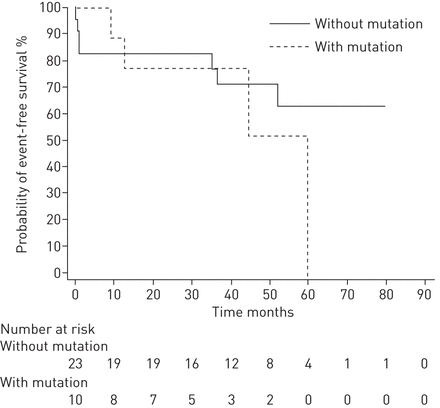
根据肺动脉高压患者突变携带者状态的无事件生存概率分布(仅限偶发病例)。考虑的事件:死亡;移植;Potts分流。使用对数秩检验比较两种分布曲线:卡方=0.6680;d.f.=1;p=0.4137。
后果
PH基因突变
通过系谱分析和临床检查,35名儿童被归类为IPAH,5名为FPAH,13名为3型APAH-CHD,10名为4型APAH-CHD,3名为PVOD。在66名儿童中的14名儿童中发现了突变:8名为IPAH患者(占IPAH的23%),4名为FPAH患者(占FPAH的80%),2名为PVOD患者(占PVOD的67%).基因没有突变BMPR2和ACVRL1在3型和4型APAH-CHD的儿童中鉴定基因(表3).
五个BMPR2(三IPAH,二FPAH),四ACVRL1(三个IPAH和一个FPAH),三个TBX4(两个ipah和一个fpah)和两个双层EIF2AK4鉴定突变(两种PVOO)(表3).
先前已经描述了十种突变,并将其归类为有害突变。已经鉴定出四种新的突变:一种是基因的大规模重排BMPR2(2-7外显子缺失)和3TBX4突变均为非义突变(补充表S2中描述了所有突变)。
未发现基因突变Eng,KCNK3.或SMAD9基因。这四个ACVRL1突变携带者表现出HHT症状。在14例已确定突变的指数病例中,对11例进行了父母基因筛查。在两个IPAH案例中BMPR2突变的发现是从头突变。其余的突变是遗传性的,在所有情况下,携带突变的父母对PAH无症状,或对PAH有单一等位基因携带者EIF2AK4PVOD的突变。
在三个家庭中TBX4突变,突变是遗传的。携带突变的父母在超声心动图中无肺动脉高压(PAP)症状,但有骨异常相关的症状TBX4突变。
突变EIF2AK4在两名患者中鉴定基因(先前在E中报告伊里斯等等。[16]),有PVOD的临床、血流动力学及胸部CT表现,无PVODEIF2AK4在IPAH或FPAH患儿中发现突变。一名8岁女童在诊断时胸部CT疑似PVOD,但未发现突变EIF2AK4随后的CT扫描、肺活检和进化证实了诊断,她的临床状况需要在随访期间进行移植。
IPAH和FPAH患者诊断时的临床状况
在诊断时,IPAH和FPAH患者如果有一个PAH基因突变,其功能较差,更常伴有右室功能障碍(右房压高)的相关体征,更常出现晕厥(表1).虽然统计学上没有不同的平均PAP,PVRI和心脏指数,但这些参数的值往往在突变载体中更差。只有一个TBX4根据Sitbon标准,突变携带者被认为是急性血管舒张试验的应答者[26]因此,前期联合治疗(内皮素受体拮抗剂和磷酸二酯酶5型抑制剂的口服联合治疗)在这一组中,使用前列腺素类似物或三联疗法作为一线治疗更为频繁。值得注意的是,在成年人群中观察到女性占优势,但突变携带者和非携带者的性别比例相似。这两组患者的诊断年龄没有差异。
两组中更严重的临床和血流动力学状态在2岁以下的儿童中占多数(4例突变,8例无突变)。
表2显示每种突变类型患者的基线特征。
讨论
本研究研究了具有各种形式的pH群体的群体的遗传状态,以更好地定义儿童的PAH的遗传建筑,并评估与不同基因突变相关的临床病症。包括IPAH,FPAH,PVOD和3型和4APAH-CHD的患者,并筛选已知的PAH和PVOD基因中的突变,包括最近鉴定的基因(TBX4,KCNK3和EIF2AK4).PAH是系统性疾病的一部分或与染色体异常相关的患者不包括在研究中。
PAH中没有突变与CHD(3 APAH-CHD)和在术后PAH(型APAH-CHD)中巧合BMPR2而且ACVRL1发现基因,表明遗传背景与iPAH不同。在这两组中,没有易突变的突变表明,血管动力学因素足以诱导PAH,或者其他尚未识别的遗传因素参与该设置。这种尺寸的样品具有94%的功率(数据未显示),以检测如果遗传背景相同测试的基因中的任何敏感性变体。以前公布的结果在一小系列APAH-CHD患者上没有检测到任何致病性突变BMPR2或ACVRL1基因;仅错义VU(根据研究,大多数VU可能是良性的生物信息学工具),但在相同的情况下未发现有害突变[18,20.,24].因此,无论是这项研究的结果还是那些已经发表的结果,都不能证明用这项研究中使用的基因来筛查这些患者是正确的。
在FPAH,BMPR2在不到一半的指数病例中发现了突变,而BMPR2在80%以上的成人家族性病例中发现了突变[27]其他的基因突变也被发现ACVRL1和TBX4这代表了在儿童中发现的近50%的突变BMPR2在成人多环芳烃人群中发现的突变比例相似,在成人中也发现了突变ACVRL1和TBX4基因的频率是BMPR2如果一起考虑,突变。该研究缺乏统计能力,以达到成年人突变分布的显着差异,因为样品尺寸很小;需要进一步的研究来确认这些差异。ACVRL1本研究中发现的错义突变已经在HHT患者中报道过,并且在受影响儿童或携带者父母中出现HHT症状的家庭中的儿童中发现过,因为PAH可能先于幼儿中的其他HHT症状,但通常存在于携带者父母中[10].事实上,Girerd等等。[10]以前的研究表明,在成人中,多环芳烃的发病年龄与基因突变有关ACVRL1早于年BMPR2突变携带者的发病年龄比未检测到突变的成年人更早。
TBX4在我们的研究中,3/40(7.5%)的IPAH/FPAH患者检测到突变,这一比例低于K厄斯特金斯-F雷德里克斯等等。[15]在哪里TBX4在21%的PAH儿童中检测到突变,没有发育异常。在儿童和成人PAH中对该基因进行常规检测将有助于确定基因突变的重要性TBX4.
TBX4突变是由患有典型的小髌骨综合征的骨骼畸形但没有PAH症状的父母传播的TBX4该基因很少引起成人多环芳烃(根据K厄斯特金斯-F雷德里克斯等等。[15])。
目前,肺部渗透的主要变化TBX4从一代到下一代的突变和在一些儿童中观察到的PAH的严重性无法解释。与亲本遗传的遗传因素的相互作用和/或表观遗传机制的参与是可能的假设,需要进一步研究。没有成年人受多环芳烃影响和携带TBX4突变可能是因为这种类型的儿童发病PAH的死亡率高,但目前没有随访和家庭数据可以支持这种假设。我们有限的经验(n = 73)在常规搜索中TBX4成人PAH患者的突变显示出类似于已经报告的突变的频率(数据未显示)[15].
三名患有PVOD的儿童临床病情迅速恶化,并接受了肺移植。虽然这三名患者有类似的演变导致肺移植,但没有发生基因突变EIF2AK4该基因在三名没有受影响兄弟姐妹的患者中发现。E伊里斯等等。[16],但其他不明遗传因素可能与该患者有关。
在这项研究中,突变携带者在诊断时有更严重的临床表现,这与先前报道的成年癌症患者的经验一致BMPR2和ACVRL1突变[7.,10].然而,关于血液动力学状况的差异没有达到意义,可能是因为涉及少数患者。由于这种情况下诊断较差的临床介绍,初始治疗在该组中更具侵略性(表2)但这种差异在结果中没有更重要(表4).有趣的是,在成人和儿童多环芳烃中,BMPR2突变携带者对钙通道阻滞剂几乎没有反应。在本研究的一系列儿童期起病肺动脉高压中,也存在这种情况TBX4突变(表2和3.) [19,28,29].
在本研究中,突变的识别不影响初始治疗;这是在获得遗传筛选结果之前,基于通常的标准。如果排除了所有需要肺移植的PVOD患者,这是正确的。
在成人中,BMPR2和ACVRL1与突变非携带者相比,突变携带者在诊断时表现更严重,死亡时间更短[7.,10,29].本研究的样本量不允许根据突变携带者状态检测结果有任何显著差异(表4和图2)病情较严重,诊断后立即需要肺移植或Potts吻合的为PVOD患者(EIF2AK4突变)或ACVRL1在成人中也观察到的突变载体[10].在K的研究中厄斯特金斯-F雷德里克斯等等。[15],没有增加死亡率TBX4与其他儿童期发病的PAH患者相比。这项研究也是如此。
本研究涉及罕见疾病,样本量小,有一定的局限性。权力的缺乏可能解释了不同群体之间没有显著差异的原因。允许流行病例(可视为幸存者)在纳入期内改变突变携带者的频率。然而,突变携带频率在流行中是相似的(2/7;29%)和意外事件(10/33;30%)。这与APAH无关,因为在整个组中没有检测到突变。虽然不太可能,但由于一些患者没有被纳入研究,或由于在纳入研究的所有患者中没有筛查某些基因,一些突变可能被遗漏了(图1).
总之,本研究表明,筛查3型和4型apah -冠心病患者BMPR2和ACVRL1这一结果证实了其他研究中未发现有害突变[18,20.,24].发生突变的PAH患者的比例与成人相当,但涉及的基因分布非常不同。TBX4突变的重要贡献指出,这些突变的外显率在成人和儿童之间是不同的。
最后,突变患者诊断的更严重的临床状况似乎与先前在成年人群中观察到的患者的诊断。
本研究中的系列研究不允许对突变儿童群体的初始治疗提出具体建议。在我们的研究中,除以下情况外,其中一个基因突变的存在并不能预测临床结果:EIF2AK4在大量儿童IPAH和FPAH患者中筛查这些突变可能有助于进一步确定这些突变对临床结果的预测价值。
儿科多环芳烃遗传诊断的一个关键问题是是否有可能检测亲属的携带者或非携带者状态,如在PVOD作为隐性疾病传播的情况下检查兄弟姐妹。早期诊断和仔细随访的好处应该超过在症状出现之前就对严重疾病作出诊断的偏见,因为症状出现的可能性尚未被准确确定。对父母来说,知道某些形式的儿科高危害性pah的预后不良将是一个高情绪压力的来源,但这将有助于他们做出为人父母的决定。在这些情况下,遗传学家和遗传顾问提供的遗传咨询应有助于父母清楚地了解遗传情况,并为他们的决策提供信息。
确认
我们感谢法国巴黎公共卫生援助组织GH Pitié-Salpètrière遗传部的Anne Leroy、Marie Christine Waill和Salim Bakas对患者进行基因分型。我们感谢法国巴黎UMR_S 1166-ICAN心脏代谢和营养研究所INSERM的David Tregouët提供统计数据分析方面的建议,以及法国巴黎Hôpital Europeéen Georges Pompidou AP-HP血液学系的David Smadja教授测量循环内皮细胞。
作者贡献:M. Levy, D. Bonnet:患者的临床护理,研究设计,文章撰写。一、Szezepanski:病人护理,数据收集。F.苏布里耶,M.埃里斯:基因资料,研究设计,文章撰写。S. Nadaud, M. Ladouceur:数据的统计分析,文章的评论。
脚注
编辑评论EUR RESPIR J.2016;48: 987 - 989。
这篇文章有补充资料可从www.qdcxjkg.com
利益冲突:披露可与本文一起在www.qdcxjkg.com
- 收到2016年1月28日。
- 认可的2016年6月7日。
- 版权所有©ERS 2016











{kind=link}
{kind=link}
{kind=link}
{kind=link}