





文摘
在本文中,我们概述知识的当前状态之间的平衡胶原蛋白生产和退化特发性肺纤维化(IPF)。金属蛋白酶与IPF的特异表达作用可能在IPF发病机理中发挥核心作用。因此,抑制金属蛋白酶在IPF可能有治疗潜力,但我们的知识的病理生理作用是受到有限的动物模型和缺乏具体的抑制剂。
肺纤维化:不同机制与纤维化的端点
弥漫性间质性肺病(ILD)的特点是不同程度的炎症和纤维化导致错乱的肺的气体交换单元。这些疾病的一个特点是胶原蛋白的异常沉积。许多ILDs已知的病因学,即。暴露于有机(如。农民肺)或无机(如。由药物引起石棉肺)粒子,(如。胺碘酮),或者与风湿病疾病有关,如系统性硬化和风湿性关节炎。大约一半的ILDs原因不明,被归类为特发性间质性肺炎(1]。到目前为止最常见的是特发性肺纤维化(IPF),具有比许多癌症的预后。
目前的证据表明,IPF的结果异常应对目前身份不明的肺泡上皮损伤。伤口愈合理论推测,IPF的结果异常响应多个微观站点的肺泡上皮细胞损伤和激活(图1)。这被认为是导致持续异常上皮修复,促进成纤维细胞增殖,逐步生成网状激活成纤维细胞和胶原蛋白的重组肺架构(2]。除此之外,有增加上皮细胞凋亡和细胞损失,尤其是相邻纤维母细胞疫源地。在IPF,异常激活肺泡上皮细胞合成几乎所有,即使不是全部,引发和维持纤维化反应的介质,可能通过一个双向异常上皮和间质细胞之间的沟通3]。Fibroblast-type细胞也出现招聘的纤维细胞循环,可能,epithelial-mesenchymal转换的过程4]。
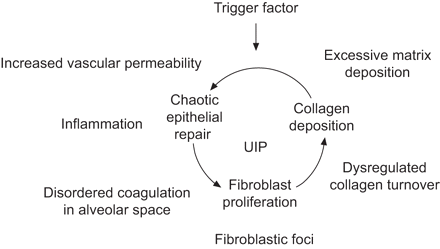
间质性肺纤维化机制(IPF)。IPF的病因学是不确定的。假设是,无论触发IPF导致上皮损伤和顺向上皮激活导致的核心特性通常的间质性肺炎(摘要),即混沌上皮修复,成纤维细胞增殖和胶原蛋白沉积,成为自我。
除了myofibroblast集中形成和上皮细胞损伤,炎症的证据有变量,就是明证增加巨噬细胞和中性粒细胞计数5),intra-alveolar凝血病(6)和新血管的形成在IPF肺(7]。异常的血管生成而且一直与肺纤维化疾病的发展(7]。在一起,这些变化导致增加肺泡毛细血管的渗透性屏障,可以检测到临床,增加二三胺penta-acetic酸间隙(8]。增加肺泡毛细血管屏障通透性也可能与早期死亡率IPF (9]。
因此,至少存在两个不同的细胞途径——炎症通路,由大多数ILDs和上皮细胞通路,如IPF——导致肺纤维化的发展10]。在本文中,我们将专注于金属蛋白酶在IPF的病理生理学的重要性。
胶原蛋白和IPF的病理生理学
有许多不同种类的胶原蛋白,但第三类型我和健康和纤维化肺内占主导地位11]。纤维胶原蛋白分泌是可溶性前驱(轴承大扩展他们的氨基和羧基末端前肽),自我形成不溶性三重螺旋纤维联系起来。胶原原纤维呈现的三重螺旋构象分子抵抗大多数酶蛋白水解攻击除了金属蛋白酶,生物学已经在本系列的第一篇文章概述12]。
大量证据存在I型和III胶原蛋白生产是IPF的增加。大多数研究关注I型或第三生产看着胶原carboxy-terminal前肽(分别PICP和PIIICP)。这些都是作为代理标记增加胶原蛋白的生产,因为胶原蛋白本身是不溶性的,因此不能直接侵入性活检采样。
已发现在IPF PICP和PIIICP高架在支气管肺泡灌洗液(BALF),但不是血清,病人。PICP水平BALF和上皮衬里液有显著负相关的肺一氧化碳扩散能力的单位肺泡体积(13]。在免疫组织化学或原位信使rna研究肺组织、III型胶原蛋白主要在肺泡隔增厚和间质,而I型胶原蛋白似乎是主要的胶原蛋白在疾病后期课程(14]。I型胶原主要是作为细胞内的点在新成立的纤维化通常的间质性肺炎(摘要),而III型胶原表达细胞外地下面化生的肺泡上皮细胞(15]。增加其他成分的细胞外基质,包括类型V, VI,七世胶原、纤连蛋白、弹性蛋白、蛋白聚糖也存在(10]。
ILD的趋向下降的环境
高浓度的胶原产品不一定等同于胶原沉积增加,因为胶原蛋白降解是一个动态的过程受基质金属蛋白酶(MMPs)及其抑制剂。为了评估净胶原蛋白沉积在肺,是否还需要胶原蛋白降解的一些评估。
一些早期的研究指出纤维化肺内胶原蛋白降解的异常。1979年,使用zymographic方法,G各方et al。(16]证明肺匀浆中胶原酶活性升高15 21 IPF患者,但在没有正常对照组或结节病病人。相反,有证据表明collagenolysis降低过敏性肺炎,在动物身上实验矽肺和bleomycin-induced肺纤维化(17]。此外,免疫组织化学研究表明高水平的表达组织金属蛋白酶抑制剂(TIMPs) IPF肺内。TIMP-1发现间质巨噬细胞和纤维母细胞TIMP-2疫源地。TIMP-3显示强烈的染色主要装饰血管的弹性板。TIMP-4表示在IPF肺上皮细胞和浆细胞(18]。另外米ontano等。(19)发现,胶原酶抑制活性更高的IPF患者的活检样本和过敏性肺炎比对照组。鉴于IPF tissue-derived肺成纤维细胞表达profibrotic分泌表型(减少胶原酶和TIMP表达升高),这些早期的研究表明,nondegrading纤维胶原微环境可能战胜ILD [18- - - - - -20.]。
在最初,缺陷collagenolysis被怀疑导致过度的细胞外基质沉积在肺纤维化,这种观点现在看来过于简单。几项研究已经表明,基质金属蛋白酶的增加,而不是在IPF(基质金属蛋白酶,图2)[9]。金属蛋白酶- 1水平升高,MMP-2 MMP-3, MMP-7, MMP-8和MMP-9报道。MMP-12和MMP-13也被卷入实验性肝纤维化。他们的角色和潜在的意义进行了讨论。
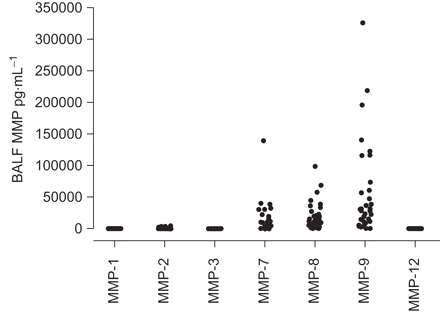
支气管肺泡灌洗液(BALF)基质金属蛋白酶(MMP)水平用散点图表示。BALF从基线和随访数据结合支气管镜检查(n = 28)。散点图表明,大多数BALF MMP的蛋白质是MMP-7, MMP-8 MMP-9。BALF MMP-2, MMP-3、MMP-7 MMP-8和MMP-9与正常对照组相比显著升高。从[复制9]。
金属蛋白酶- 1
金属蛋白酶- 1已被证明是高一些,但不是全部,IPF患者BALF研究,一项研究表明等离子体水平也增加了(9,21]。微阵列数据表明,金属蛋白酶- 1 mRNA显著调节整个肺组织从IPF患者与过敏性肺炎(22)以及正常控制肺(23]。金属蛋白酶- 1的表达也是更高的家族与零星的IPF患者(24]。有趣的是,在金属蛋白酶- 1基因启动子多态性与IPF更常见于吸烟者,揭示一个假定的基因-环境相互作用在这种疾病25]。
IPF的观察金属蛋白酶- 1是调节,一个条件与积累的我和III型胶原,乍一看是一个悖论,特别是当它也被牵连在慢性阻塞性肺疾病的发病机制,失去弹性组织的一个特性。一个可能的解释是,金属蛋白酶- 1的表达主要是反应性上皮,不是在胶原蛋白积累的间隙间10]。另外,金属蛋白酶- 1的活性可能会抵消组织抑制剂,导致只有弱的活动。然而,金属蛋白酶- 1的生物学作用,除了胶原降解,包括细胞因子的处理,如pro-tumour坏死因子(TNF) -α,调节细胞迁移,和潜在的细胞生长26]。这些金属蛋白酶- 1的多个生物功能,随着临床数据,建议IPF发病机制中的一个重要的角色。
MMP-2
MMP-2(白明胶酶)据报道,广泛表达于肺纤维化,特别是覆盖intra-alveolar纤维化增生的上皮细胞、成纤维细胞和间充质细胞病灶(18]。MMP-2已经报道,在BALF升高,但在另一项研究中,西方的屁股主动MMP-2建议只弱活动组织(特别是与闭塞性细支气管炎肺炎(BOOP) () (9,27]。MMP-2降解多种矩阵和nonmatrix基质,尤其是IV型胶原蛋白和其他基底膜蛋白质。此外,MMP-2通常是调节实验模型的肺纤维化及其超表达,以及MMP-9,被怀疑与基底膜破坏(28]。这可能是重要的,因为肺泡壁的结构完整性取决于基底膜和认可,牙龈的破坏基底膜可能会先于肺泡纤维化的发展。一个不连续的基底膜可能允许更大的访问渗出性肺泡空间因素和间质细胞,进一步促进组织破坏和进行性纤维化(9,29日]。
MMP-3
MMP-3 (stromelysin 1)水平升高在IPF患者BALF和观察到的那些死在三年的诊断9]。MMP-3可能纤维化的重要推动力,因为MMP-3转基因老鼠刺激发展的表达在上皮细胞纤维化和随后的肿瘤形成。乳腺上皮细胞进一步接触MMP-3诱发epithelial-mesenchymal过渡,细胞获得myofibroblast-like特点,这个过程是依赖于细胞活性氧的生成。数据从文化模型诱导基质金属蛋白酶的表达在人类肺癌细胞系和转基因小鼠模型中基质金属蛋白酶表达的诱导肺肺泡上皮细胞显示类似的过程可能发生在肺(30.,31日]。MMP-3也被卷入反血管增生胶原蛋白降解产物的释放,如血管内皮抑制素,能促进肺泡上皮细胞凋亡,这被认为是一个重要的驱动程序正在进行的纤维化(32,33]。
MMP-7
MMP-7,也称为matrilysin,据报道是最稳定的基因在肺纤维化的升高。MMP-7表达式不不同家族之间,零星的IPF (34,35]。IPF肺,增加主要由肺泡上皮细胞异常免疫反应性的蛋白质表达和活性蛋白质已经证明了组织zymography在IPF的肺34]。在BALF, MMP-7水平与肺功能损害的严重程度在IPF (9]。最近,它还表明,高浓度的MMP-7也可以发现在非特异性间质性肺炎和结节病36),这表明增加不是特定于IPF MMP-7表达式。
MMP-7被描述为一个profibrotic金属蛋白酶(10]。几行研究表明MMP-7可能促进纤维化反应通过监管影响上皮修复和释放潜在转化生长因子(TGF) -β。MMP-7零老鼠相对免受bleomycin-induced纤维化,表明这个MMP的核心驱动组织纤维化反应。MMP-7具有广泛的底物的亲和力细胞外基质成分,包括IV型胶原、层粘连蛋白、纤连蛋白、明胶、弹性蛋白和骨桥蛋白。MMP-7也有能力来处理许多生物活性基质,如Fas配体(FasL),β4整合素,钙粘蛋白、pro-TNF-αpro-α-defensin、血管内皮抑制素syndecan和α2巨球蛋白。MMP-7也可以激活蛋白酶包括本身和pro-MMP-1 pro-MMP-2 pro-MMP-9。从细胞外基质释放成品TGF-βMMP-7 TGF生物活性的主要监管机构,这可能促进纤维母细胞生长,生存和胶原蛋白合成。因此,MMP-7在肺纤维化中的作用可能是多向性的,由于其多样化的生物作用,参与细胞凋亡、炎症、fibroproliferation和先天免疫(10]。
MMP-8
MMP-8 (collagenase-2或中性粒细胞胶原酶)来源于中性粒细胞,并在较小程度上,从成纤维细胞和内皮细胞、上皮细胞和浆细胞。MMP-8水平升高在IPF患者BALF和与BALF的溶胶原的能力(37]。MMP-8 IPF患者BALF的最高水平在快速进行性疾病和贫穷的生存,和肺泡水平不降低联合治疗(强的松、咪唑硫嘌呤和N乙酰半胱氨酸(NAC)) (9]。因此,似乎MMP-8水平升高与IPF的不良结局。
中性IPF患者的牙槽炎是一个特性和嗜中性的程度一直与BALF死亡率在一个大系列(5),所以它是有趣的推测,嗜中性驱动矩阵营业额通过MMP-8,至少在一些病人。另外,MMP-8已经涉及纤维细胞的迁移和归航。纤维细胞是独一无二的骨骨髓来源间充质祖细胞,是由他们的生长特性和表面表型,因为他们表达标记与白细胞兼容,造血祖细胞和成纤维细胞。纤维细胞的肺内发现了IPF患者预后和循环纤维细胞水平低下的一个标志在IPF (38,39]。衰减的纤维细胞也贩卖小鼠模型直接与肺纤维化的减少。显然,潜在MMP-8可能扮演重要的角色在这个过程显然是重要的需要进一步研究。
MMP-9
MMP-9(白明胶酶B)已被广泛研究在IPF患者主要是表达的肺泡巨噬细胞、嗜中性粒细胞和上皮细胞。正常的肺,MMP-9不是由居民细胞产生,但在各种形式的刺激下,支气管上皮细胞,克拉拉细胞,肺泡II型细胞,成纤维细胞、平滑肌细胞和内皮细胞可以产生MMP-9 [40]。MMP-9基因表达和蛋白质被发现在人类和高架实验性肺纤维化(41,42]。成纤维细胞和肺泡巨噬细胞提取IPF患者肺部产生MMP-9升高与正常细胞相比,(19,41]。
MMP-9水平BALF和MMP-9活动在迅速进步的IPF病例的样本(最大9,43]。海拔的MMP-9是否激活中性粒细胞的标志或参与这个子集的肺泡损伤病人是未知的。然而,海拔MMP-9 (BOOP)患者BALF中超过在IPF (44与中性粒细胞),这表明一个协会而不是肺组织学。
动物研究使用MMP-9基因敲除小鼠显示一些相互矛盾的结果这个金属蛋白酶的作用。博来霉素后安装,MMP-9-null老鼠开发纤维化,类似于由野生动物,尽管MMP-9-deficient小鼠的肺显示肺泡bronchiolisation最小,这表明MMP-9促进迁移的克拉拉细胞和其他细支气管肺泡损伤的区域(45]。另外,过度的MMP-9巨噬细胞已被证明会减弱bleomycin-induced纤维化(46]。减少profibrotic介质,如TIMP-1和胰岛素样生长因子结合蛋白(IGFBP) 3、在MMP-9转基因老鼠被确认为一个潜在的减少纤维化反应的机制。很难调和这些发现,但他们建议MMP-9可能促进或降低纤维化反应。似乎总体响应取决于细胞产生MMP-9,当地组织抑制剂水平和可用的目标分子/基质。
MMP-12
MMP-12(巨噬细胞弹性蛋白酶)与纤维的反应在动物实验中使用FasL的纤维化。老鼠对Fas-activating抗体增加了caspase-3激活肺泡壁细胞和肺总胶原蛋白增加曝光后21天。基因表达分析显示顺序激活粘住profibrotic基因,包括MMP-12 upregulation标志。目标删除MMP-12保护小鼠免受Fas-induced肺纤维化,即使肺部的炎症反应是类似于野生型小鼠(47]。
很少有数据MMP-12 IPF患者或表达水平。在一个小型研究,MMP-12只是检测BALF中三18 IPF患者和代表只有0.022%的总IPF BALF MMP的水平,以Luminex array [48]。重要的是要认识到,BALF MMP-12水平不一定清楚地反映组织水平由于当地复杂的调节基质金属蛋白酶抑制剂。因此,现有的证据不支持MMP-12起到了很大的作用在人类肺纤维化与IPF有关。
MMP-13
MMP-13一直参与炎症和纤维化的严重程度在实验asbestos-induced肺损伤,随着MMP-2, MMP-9 MMP-12。使用通用MMP抑制剂,GM6001、减毒的炎症和纤维化反应的程度49]。MMP-13基因敲除小鼠也有减少急性炎症和纤维化当暴露于辐射50]。
相比之下,在肺纤维化和百草枯诱导大鼠氧过多,Ruiz等。(28]证明了降低胶原酶水平MMP-8和MMP-13 TIMP-1和TGF-β的增加。因此,类似于前面描述的MMP-9动物模型的结果,结果MMP-13是相互矛盾的。对人类IPF MMP-13的角色,尽管MMP-13 IPF BALF中检测不到(湄Thickett,未发表的观察)。这些数据支持免疫组织化学和rt - pcr IPF肺(18]。
在IPF膜相关和其他金属蛋白酶
基质金属蛋白酶的一个子集,膜式(MT)基质金属蛋白酶的激活参与pro-MMP-2 MMP-2。这些MT-MMPs已被证明在IPF的肺部组织。MT1-MMP MT2-MMP在肺泡上皮细胞被发现,MT3-MMP从成纤维细胞的疫源地和肺泡上皮细胞、成纤维细胞和基底细支气管上皮细胞和MT5-MMP鳞状上皮化生。肺成纤维细胞,TGF-β1诱导强烈的MT3-MMP upregulation,基因和蛋白质水平(51]。
已知MMP的生物多样性的增加意味着许多潜在的重要基质金属蛋白酶在IPF学习仍然不佳。例如,微阵列研究表明高MMP-10 MMP-28 IPF的组织,但他们的细胞来源,基质的特征和功能不佳。MMP-28可能特别感兴趣,因为它提出了一个角色在上皮间充质转化和蛋白水解乳沟TGF-β[52]。进一步研究阐明这些新的基质金属蛋白酶的重要性正在进行。
目前的治疗效果IPF在肺MMP的表达
目前治疗IPF仍不满意,几乎没有证据表明,纤维化的摘要模式与治疗肺活检曾经就退化。最近的一次试验,然而,建议用强的松治疗,用药和南汽放缓发展(25),但目前还不清楚这种治疗减少异常的胶原蛋白流失。唯一的预处理和后处理研究解决MMP的水平在同一个人证明,结合药物治疗没有任何抑制活动在BALF MMP的免疫反应性(9]。这尽管是一个先前的研究表明,类固醇治疗减少MMP-9生产IPF患者(26];然而,在这个研究中,患者没有单独研究连续预处理和后处理。
Pirfenidone是小说antifibrotic代理已经被证明可以减少胶原蛋白沉积在各种动物模型在活的有机体内。试验在IPF患者(53)表明,它减少了肺功能的下降速度,导致最近的欧洲药品局建议pirfenidone被许可用于IPF在欧洲。尽管pirfenidone的作用机制尚不清楚,临床前研究表明,调制发表的几MMP活性成分。在肝纤维化模型中,pirfenidone antifibrotic效应主要是由于原骨胶原的表达式和TIMP-1 TGF-β1的差别很可能通过对这些基因的信使rna, MMP-2 [54]。在小鼠气管内的脂多糖,pirfenidone减少MMP-9由于中性粒细胞减少招聘55),而低剂量pirfenidone抑制TGF-β1 TIMP-1,博来霉素引起的肺纤维化和保护老鼠,但对MMP-13[的表达没有影响56]。
金属蛋白酶IPF的生物标志物
IPF的金属蛋白酶含量增加肺的发现和报告关系严重的肺功能下降或进行性疾病BALF导致兴趣是否有潜力作为生物标志物面板用于IPF基质金属蛋白酶。BALF生物标志物将使用有限的物流支气管肺泡灌洗,变量稀释的效果和广泛的生物变异个体MMP的水平。
最近,一组49血浆蛋白测量等离子体的IPF患者从控件定义一个five-protein签名,杰出的病人。MMP-7和金属蛋白酶- 1,血浆蛋白的水平是两个最增加IPF患者与控制相比,都是关键的组件的签名(MMP-7,金属蛋白酶- 1、MMP-8 IGFBP1和肿瘤坏死因子受体超家族成员1 a)。面板是足以区分病人和控制的灵敏度为98.6%。这些结果进一步验证了在一个独立的验证IPF患者群,家族性肺纤维化和亚临床ILD和控制个人(57]。鉴于这些血浆蛋白可以以单个Luminex试验,这个面板标记有一些潜在的,特别是水平升高的患者亚临床疾病被高分辨率计算机断层扫描。然而,这个小组和疾病进展之间的关系并没有报道。我们需要在临床实践中比一个诊断疾病的生物标志物的活动面板。
金属蛋白酶IPF的治疗靶点
基质金属蛋白酶的upregulation癌症和炎症性疾病药物研发让他们有吸引力的目标在过去的20年。我们才能期望MMP抑制剂在IPF患者是有效的,过量的胶原蛋白沉积特征病理发现吗?
增加识别金属蛋白酶的生物功能的复杂性伤口修复、血管生成、及其影响细胞因子,趋化因子和生长因子释放表明有可能抑制调节异常的肺泡IPF改造。Bleomycin-induced肺纤维化是由非特异性MMP抑制剂减毒的行动的强力霉素抗生素58]。这一行动与减少肺部炎症和降低BALF (MMP的活动59]。此外,一个小型的开放研究的强力霉素治疗七IPF患者没有文档用力肺活量下降尽管>日常使用强力霉素的17个月。这样一个缺乏进展将是不寻常的在一个典型的IPF患者队列,但研究的质量不允许任何结论是IPF患者(强力霉素治疗的功效60]。
金属蛋白酶抑制的热情也必须受到使用广谱抑制剂在癌症早期试验的失败,以及潜在的不利影响,减少治疗的担忧胶原降解fibrosing疾病的可能。理想情况下,MMP抑制剂用于IPF会为个人基质金属蛋白酶特异性。研究人员在这一领域面临的挑战是,因此,识别任何个人MMP是否IPF的关键机械的司机。这样的工作由依赖模型,阻碍bleomycin-induced损伤等无法正常人类疾病模型(61年]。
结束语
IPF是一种毁灭性的疾病,和当前/新兴治疗不满意是因为毒性和有限的功效。目前的IPF发病机理的理论表明,肺泡上皮损伤引起的间充质细胞与成纤维细胞的迁移和增殖形成焦点。病态,这导致夸大了胶原蛋白沉积在肺的某些部分,导致上皮结构和蜂窝的形成。尽管看到进步的疤痕,证据,增强金属蛋白酶的生产。这些酶的作用目前还不清楚,因为他们有多效性的影响和细胞外基质处理的趋化因子、细胞因子和生长因子。可能是调节矩阵退化,因此,机械的司机IPF的进行性纤维化。在这个领域的研究是阻碍了IPF的缺乏良好的动物模型,但更好的理解IPF的病理生理学和胶原蛋白营业额应该确定新的治疗目标这个毁灭性的疾病。
脚注
本系列之前的文章:1号:Loffek年代,先令O, Franzke北京市。生物基质金属蛋白酶的作用:一个关键的平衡。欧元和J2011;38:191 - 208。2号:Elkington PT, Ugarte-Gil CA, Friedland JS。基质金属蛋白酶在肺结核。欧元和J2011;38:456 - 464。3号:Gaggar,赫克托耳,Bratcher PE、et al。基质金属蛋白酶的作用在囊性纤维化肺病。欧元和J2011;38:721 - 727。4号:阿,戴维,McAuley DF 'Kane厘米。基质金属蛋白酶在急性肺损伤:损伤介质和司机的修复。欧元和J2011;38:959 - 970。5号:Vandenbroucke再保险,Dejonckheere R, Libert c .基质金属蛋白酶抑制剂在肺部疾病的治疗作用?欧元和J2011;38:1200 - 1214。
感兴趣的语句
一份声明中对湄Thickett可以发现www.www.qdcxjkg.com/site/misc/statements.xhtml
- 收到了2011年2月9日。
- 接受2011年4月30日。
- ©2011人队











{kind=link}
{kind=link}
![Bronchoalveolar lavage fluid (BALF) matrix metalloproteinase (MMP) levels expressed as a scatter plot. BALF data were combined from baseline and follow-up bronchoscopy (n=28). The scatter plot demonstrates that the majority of BALF MMP protein is MMP-7, MMP-8 and MMP-9. BALF levels of MMP-2, MMP-3, MMP-7, MMP-8 and MMP-9 were significantly elevated compared with normal controls. Reproduced from [9].](http://www.qdcxjkg.com/content/erj/38/6/1461/F2.large.jpg?width=800&height=600&carousel=1){kind=link}
{kind=link}