





文摘
Pleuroparenchymal fibroelastosis (PPFE)是一种罕见的疾病的特点是主要上部叶胸膜和在底下的实质纤维化,后者intra-alveolar伴随肺泡壁的弹性组织变性。本研究的目的是审查情况下满足成像和组织学标准出版,并识别任何常见的临床特征,可能意味着一个潜在的病因学为条件,曾被认为是特发性。
12例(7个女性,平均年龄57岁),表现症状是气短12例(11)和干咳12例(6)。七个病人复发感染过程中报道他们的疾病。5显示非特异性自身抗体积极性。两个病人有家族史的间质性肺病(ILD)。
高分辨率计算机断层扫描特性的肺部疾病远离pleuroparenchymal变化出现在六12例(同时共存的纤维化,n = 5;支气管扩张,n = 1)。七患者组织抽样从叶越低,四个病人显示那么激烈PPFE变化(过敏性肺炎的一个附加功能)和三个显示常用的间质性肺炎。
PPFE是一个独特的临床病理的实体,与临床数据表明反复肺部感染的链接。遗传和自身免疫机制也可能导致这些变化的发展。PPFE也可能存在扩散比先前报道的参与,和共存ILD的不同模式。
Pleuroparenchymal fibroelastosis (PPFE)是一种罕见的疾病,首先日本文学中所描述的米塔尼et al。(1]上层叶特发性肺纤维化。PPFE由致密建立intra-alveolar纤维化,肺泡壁弹性组织变性在这些领域表现突出,和致密纤维肺胸膜增厚;这些变化有一个引人注目的上层优势(2,3]。所知甚少关于其病因学和大多数情况下被认为是特发性,尽管几例家族性(1,2,4- - - - - -6)和其他报告与先前的骨髓移植协会(7]。此外,英语中大多数报告迟签2002年美国胸科学会(ATS) /欧洲呼吸学会(ERS)共识特发性间质性肺炎的分类(188bet官网地址8),所以没有统一定义关于PPFE诊断标准,特别是是否应该被视为一个真正的特发性实体。
因此,本研究的目的是评估患者的病理和放射性的发现实现PPFE标准出版和审查临床数据对可能的病因学。
方法
诊断病理学皇家档案主管布朗普顿和地铁站Harefield NHS信托基金会(英国伦敦)寻找案件中,术语“intra-alveolar纤维化”,“pleuroparenchymal”和“fibroelastosis”。33个病例和显微镜载玻片从30例综述了由两个病理学家(A.G.尼科尔森和j . von der Thusen)使用出版的组织学诊断标准PPFE [2,3]。这些随后被以共识为“定PPFE”、“PPFE一致”或“不符合PPFE”。“定”被分配当有上层区胸膜纤维化在底下的intra-alveolar纤维化伴有肺泡隔弹性组织变性。“符合”被分配当intra-alveolar纤维化在场但它不是1)与重要的胸膜纤维化,2)主要胸膜下或3)上层叶活检。“不符合”被分配上面描述的情况下,缺乏必要的特性。30个病人的组织材料,21患者高分辨率计算机断层扫描(HRCT)图像供审查,这是由两个放射科医生(D.M. Hansell和T.L. Reddy)。这些都是独立特征显微镜样品以类似的方式,根据报道HRCT特征从先前的案例研究,组织学证实[2,3]。“定”被分配为例,演示了胸膜增厚与胸膜下纤维化相关集中在上部叶,叶不明显低的参与或缺席。“符合”被分配情况下,上部叶胸膜增厚与胸膜下纤维化相关在场但1)分布的这些变化不是集中在上部叶或2)有功能的同时共存的疾病。疾病在其他叶记录按照2002 at /特发性间质性肺炎的分类和2011人队共识ATS /人/日本呼吸协会/ Asociacion上面de Torax循证指南特发性肺纤维化的诊断和管理(8,9]。“不符合”被分配上面描述的情况下,缺乏必要的特性。例仅包括当他们被认为是明确的或符合PPFE组织病理学和放射学。如果病理学家或放射科医生表象归入PPFE与诊断不符,被排除在外。
有17个30和14的21例明确的或符合PPFE组织病理学和成像,分别。四个病例被排除在17日接受组织审查标准由于缺乏计算机断层扫描,而额外的情况下被排除在外,因为它没有履行PPFE成像标准(不符合)。的14例hrct可用,两种情况被排除在外,因为他们没有履行组织学标准,这些案件揭示过敏性支气管肺的曲霉病(n = 1)和一个纯粹bronchocentric intra-alveolar纤维化(n = 1)。因此,12例由最后一个学习小组和临床数据对这些从病人的笔记或检索电子病历数据库。
结果
临床数据
临床资料和调查总结表1和2。的12个病人满足入选标准,七是女性,五是男性(年龄24 - 85岁,平均57岁)。表现症状包括呼吸短促(n = 11)和干咳(n = 6);诊断前症状的持续时间是6个月到6岁(平均2年8个月)。七个病人下呼吸道感染复发,都有至少有三个急性发作前诊断。三个病人发展自发气胸或纵隔气肿的过程中他们的疾病。两名患者报告暴露于环境过敏原模具和鸟类的形式;其中一个病人模具和禽流感的沉淀素检测呈阴性,其他病人没有接受测试。已知两个患者的一级亲属间质性肺病(ILD) (biopsy-proven通常的间质性肺炎(摘要),n = 1;放射检查可疑的非特异性间质性肺炎(NSIP) /摘要,n = 1)。 With the exception of one patient (immunosuppressed following a renal transplant for anti-neutrophil cytoplasmic antibody-positive glomerulonephritis), no significant drug history was evident in the study group. Specifically, there was no history of radiotherapy or chemotherapy. None of the patients had undergone bone marrow transplantation. Four of the seven patients who reported recurrent infections also demonstrated autoantibodies in their serum. One of the patients who tested positive for曲霉属真菌免疫球蛋白G抗体(表2)有一个小的焦点bronchocentric肉芽肿病叠加在PPFE肺活检组织学检查。四个五个患者自身抗体的遥远的间质纤维化的成像特性,要么出席初步审查(例2、3和8)或随后发展(例5)。一个病人被诊断与杜丝勒之前住院综合征(案例7)。9个病人支气管肺泡灌洗,一个病人表现出高水平的嗜酸性粒细胞(例3)。所有患者结核培养结果为阴性。
图像数据
计算机断层扫描发现在所有12例显示双边pleuroparenchymal不规则增厚,这是最为明显的上部和中部地区,有一个关联的胸膜下网状模式符合建立纤维化(图1)。大多数病人显示胸膜下纤维化中等程度的12例(8)。
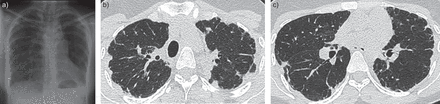
例4,35-yr-old女性。一)胸片显示两国顶端不规则胸膜增厚。有纵隔气肿和上部叶体积损失门的高度。高分辨率计算机断层扫描通过b)上和c)中间区域的典型特征pleuroparenchymal fibroelastosis,双边胸膜不规则增厚,在底下的网状模式符合纤维化。
5个12例演示了地区间质纤维化远离pleuroparenchymal变化(扣除情况3,这被认为是扩散(all-zone) PPFE成像)、纤维化的HRCT模式是最让人想起NSIP /可能的摘要(3例),NSIP(一个病人)和不可归类的间质性肺炎(1例);这些模式是集中在中下游地区(无花果2和3)。
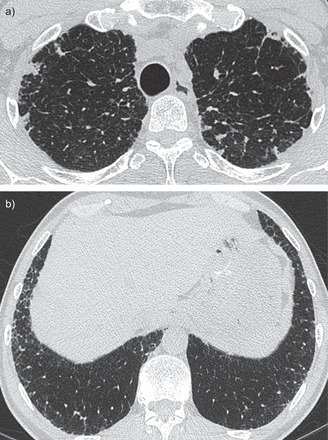
6,59-yr-old男性。)高分辨率计算机断层扫描通过上部叶,展示的特性pleuroparenchymal fibroelastosis。b)胸膜下网状低叶,符合非特异性间质性肺炎/非典型间质性肺炎。

案例8,57-yr-old女性。)高分辨率计算机断层扫描(HRCT)通过上部叶。除了功能符合pleuroparenchymal fibroelastosis,有网状模式、片状毛玻璃变化和相关的牵引在底下的支气管扩张的实质。b)通过降低叶HRCT显示广泛的细网状和毛玻璃不透明,让人想起纤维化非特异性间质性肺炎。
6个病人小病灶的整合,所有在一个上层分布。一个病人co-existent支气管扩张,潜在过敏性支气管肺的曲霉病的可能性(ABPA)在这个病人长大,HRCT的研究结果的基础上(图4)。
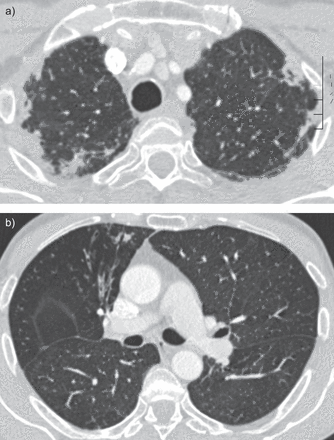
9,65岁的男性。)计算机断层扫描部分的对比度增强的研究表明特征pleuroparenchymal fibroelastosis肺的顶端。b)前节,对上部叶圆柱与粘液堵塞支气管扩张;在其他部分,有一个背景的马赛克衰减反映small-airway闭塞。
串行计算层析x射线摄影机是在六12个病人,这些病人证明了稳定或轻微进展对pleuroparenchymal变化(8-51月,平均14个月)。一间质纤维化患者表现已经证明明显纤维化的进展根据后续的计算机断层扫描进行了8个月后(8例)。一个病人没有同时共存的间质疾病首次评估了标记下叶纤维化的42个月后执行后续的计算机断层扫描(例5)。
病理数据
上部叶的肺活检在10个病人的计算机断层扫描的3个月内进行。海豆芽和较低的一个病人活检叶,与海豆芽符合PPFE显示功能,和一个病人诊断与所有叶取样做尸检。所有患者证明intra-alveolar纤维化与隔弹性组织变性(IAFE) 12例(12),11种显示胸膜纤维化(图5)。其他肺上叶变化识别包括焦点perilobular(12)的和bronchocentric(10的12)IAFE (图6)。部分肺血管的狭窄、动脉和静脉,被确认在8纤维化领域内的12例;在两种情况下,这足以促使vasculo-occlusive疾病的建议在原来的报告(图7)。肉芽肿性炎症在三个患者被确认:在两个是局部的,在一个有一个混合的局部肉芽肿和bronchocentric栅栏样的炎症(图7)认为反映叠加ABPA,尽管没有确定真菌。所有病例显示变量的非特异性慢性炎症,局部淋巴滤泡的形成。

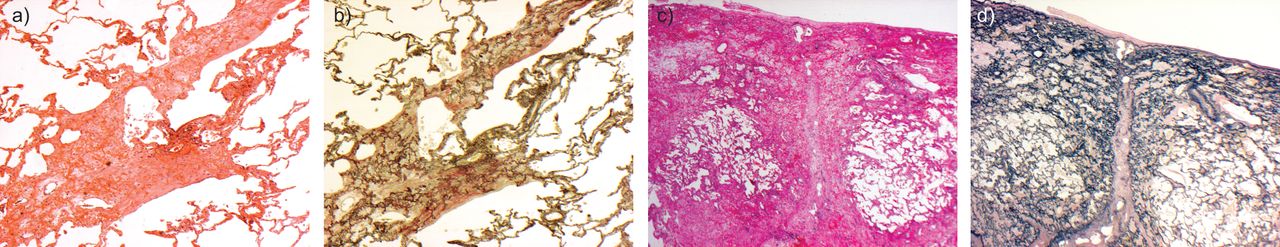
a)的pleuroparenchymal fibroelastosis显示致密胸膜下纤维化与b)著名的弹性组织变性,c)由弹性蛋白沉积在肺泡壁的网络,与干预胶原沉积。d)第二例尸检显示额外突出胸膜纤维化。苏木精和伊红染色:);罪犯)弹性Van Gieson染色。放大:a, b, d) 2×;100×c)。
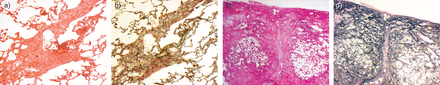
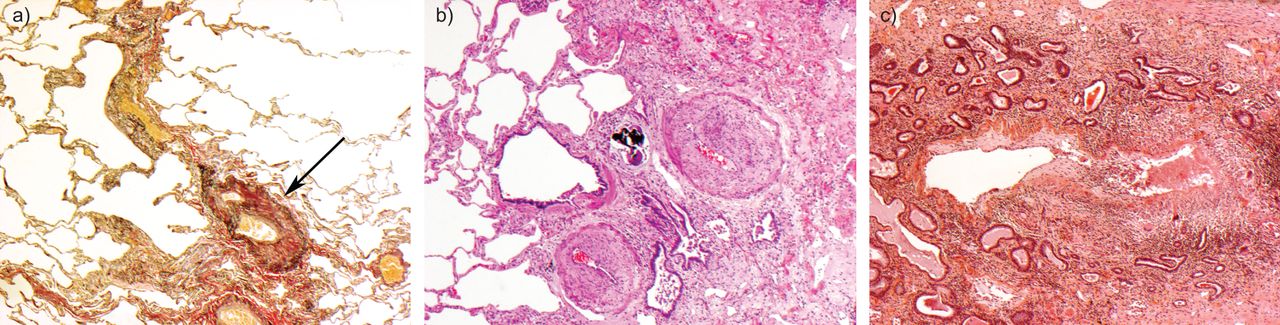
a、b) Intra-alveolar胸膜纤维化遥远显示peribronchiolar分布,延长环绕一个肺泡管(右上角)。c, d)其他领域显示了更多perilobular分布随着intra-alveolar远离胸膜纤维化延伸。染色:和,c), c)苏木精和伊红;b, d)弹性Van Gieson。放大:a, b) 100×c, d) 20×。
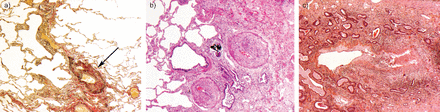
由内层的纤维化)局部静脉阻塞。b)类似的内膜的变化出现在肺动脉和包含Schaumann烧毁的肉芽肿的身体也看到毗邻细支气管。c)一个小气道显示肿物坏死性栅栏样的肉芽肿性炎症,类似于bronchocentric肉芽肿病。染色:a)弹性Van Gieson;b, c)苏木精和伊红。放大:a、b)×100;40×c)。
六个病人第二次活检,从较低的叶,1例尸检诊断也从左右两叶低采样。所有7显示额外fibrosing肺病。4例显示IAFE形态相同,虽然明显低于,变化出现在上部叶在四种情况下,摘要和3例显示模式(图8)。IAFE的患者特征之一的叶越低,有co-existent过敏性肺炎的特点包括bronchocentric组织肺炎、慢性炎症和焦一起小,差non-necrotising肉芽肿形成。值得注意的是,IAFE出现更多bronchocentric下叶切片用更少的胸膜增厚。

的结合pleuroparenchymal fibroelastosis和通常的间质性肺炎(摘要)。a、b)建立上部叶的活检显示不完整的纤维化与边缘地区的成纤维细胞的变化。弹性冯Giesen (EVG)染色显示intra-alveolar纤维化,与弹性组织变性肺泡壁的残留物。c, d)下叶相同的患者的活检显示了一筹纤维化,再次与成纤维细胞的变化,但EVG染色显示稀疏,更加分散,缺乏组织弹性蛋白染色,典型的摘要。苏木精和伊红染色:a、c);b, d) EVG。放大:40×。
Imaging-pathology相关性
3的6个患者的HRCT特征co-existent纤维化(NSIP /摘要辐射模式,n = 2;扩散PPFE辐射模式,n = 1)进行了活检和被组织学证实co-existent摘要。在HRCT上三下叶纤维化的患者特征没有下叶切片,与HRCT模式类似NSIP (n = 1), NSIP /可能的摘要(n = 1)和不可归类的间质性肺炎(n = 1)。
四个病人表现出遥远的ILD组织学上没有相应的HRCT特征异常。这四个患者的组织学模式三是PPFE,组织学上的IAFE代表(例1、9和11)。第四个病人组织学特性表明扩散PPFE和过敏性肺炎(10例)。
治疗
完整的治疗记录中可用的12例。都处理的低剂量糖皮质激素口服强的松的形式,有两个病人接受额外的高剂量皮质类固醇以脉冲的形式静脉methylprednisone。两个病人接受额外的免疫抑制剂治疗的形式环磷酰胺(n = 1)和咪唑硫嘌呤(n = 1)。两个病人治疗N乙酰半胱氨酸。一个病人接受两个(环磷酰胺)和免疫抑制剂治疗N乙酰半胱氨酸。两个病人接受预防性抗生素(阿奇霉素)复发感染。活组织检查显示疑似叠加ABPA的一个病人是抗真菌药物治疗(10例)和显示,对治疗的反应。
的10个病人随访数据,7演示了疾病进展。五个病人死了,之间的时间间隔PPFE和死亡的诊断从4个月到2年。四个五个病人复发感染过程中报道他们的疾病。五个病人阳性曲霉属真菌sp。这四个病人也有积极的自身抗体的屏幕。
两个病人的临床课程co-existent纤维化的稳定表现特征。病人与HRCT特征类似NSIP /可能的摘要进行了活组织切片检查,这是组织学证实为摘要,而非特异性的HRCT特征纤维化患者没有接受下叶活检。
讨论
本研究描述了一群病人实现出版组织病理学和成像PPFE标准。审查我们的临床资料证实,这种情况似乎是一个独特的临床病理的实体,尽管25%的人同时共存的摘要在较低的叶和其他情况下PPFE在其他区域的显示特性,在HRCT或肺活检。
英国文学(虽然很少报道2,3),PPFE更经常被日本企业(1,4- - - - - -5,10- - - - - -14];一个荟萃分析可用的文学(表5)显示了男性/女性的比例2/1,相比之下,我们的系列,有一个男/女1/1.4的比例。病人的年龄中位数从先前的研究是46岁,比在我们的系列中,年轻的平均年龄为57岁。可比的结果关于吸烟史,以前和目前的系列报道“从不”吸烟史在大多数(分别为62%和75%)。
在病因学方面,有几个观测可能相关的原因和/或协会,这通常被认为是一种特发性条件。首先,超过一半的患者复发性感染报道的过程中他们的疾病。这是高于数据报告米塔尼et al。(1)(23%)和Frankelet al。(2)(40%)。其中一个病人叠加ABPA,组织学,血清学和对治疗的反应。此外,1例拒绝与明确的病理检查,但计算机断层扫描特性在协会背景ABPA co-existent麴菌瘤,切除一直进行的(后者被排斥的原因)。Piciucchiet al。(15)最近报道一例阳性的病人曲霉属真菌沉淀素。查看这些数据,它可以推测,反复炎症损伤的易感性可能导致这种IAFE模式。局部胸膜增厚患者据报道ABPA和囊性纤维化,贷款支持这个理论(16- - - - - -19]。
其次,五个病人表现出自身抗体表明在某些病人自身免疫可能是一个因素。另一个病人曾被诊断出患有杜丝勒氏综合征,条件与自身免疫的基础上(20.- - - - - -22]。一个病人表现出高水平的自身抗体有肾移植。特征描述了PPFE骨髓移植后的患者(7),进一步支持自身免疫性因素疾病的发展。
遗传素质进一步可能是一个因素。一个zoulayet al。(6]报道双边顶端胸膜纤维化在三个兄弟姐妹,都在他一个潜在的病因学并不确定。在我们的系列中,两个ILD的12名患者有家族史,较低的频率比其他系列(出版mataniet al。(1),30%;Frankelet al。(2),40%;年代hiotaet al。(4),57%)。
这些数据点的一个原因,大多数情况下PPFE可能会继续被视为特发性。然而,我们的研究结果表明,例诊断为PPFE感染应该追究特性(特别是曲霉病)、自身免疫和家庭历史,所有这些可能会影响管理。
我们的一系列PPFE患者有一个盛行的co-existent ILD。四个患者不同的组织模式,两个(活检确诊摘要本文对计算机断层扫描co-existent间质纤维化的特点。其他四个病人表现出的成像特性下叶纤维化不接受第二个网站的活检。有趣的是要注意,下叶(摘要)纤维化进展在两种情况下串行成像的区域PPFE上部叶保持稳定。此外,4的5非特异性自身抗体的患者积极证明成像特性的遥远的纤维化过程中他们的疾病。据估计,15 - 20%的病人与慢性ILD随后有一个神秘的结缔组织疾病或开发一个临床上明显的结缔组织疾病(23)和组织学特性类似于PPFE报告顶端纤维化患者已知强直性脊柱炎(24,25]。增加自身抗体的水平也被报道患有间质性肺纤维化(IPF)的研究,尽管它已经暗示,IPF患者自身抗体的存在可能代表一个特异性的结果继发于肺部炎症和损伤(26,27]。最后,虽然不是从北美系列中描述2),co-existent纤维化,包括摘要、偶尔会被更广泛的日本文学中描述(4- - - - - -5,10,12]。综上所述,这些数据支持这一概念,病人可能有遗传倾向肺纤维化,可能表现为不同的组织病理学fibrosing肺病的模式。
在组织病理学方面,但不是计算机断层扫描的特点PPFE还发现在较低的四叶的七个病人,尽管有更大程度上bronchocentricity和疾病本质上是温和的。因此,可以认为这一成果进一步支持了这样一种观点,airway-centred损伤可能是这种疾病的发病机制的关键,正如上面所讨论的关于PPFE的病因学。
我们的观察强调一些关键概念有关病理学的集成多学科小组(联合化疗)管理的弥漫性肺部疾病患者和确保多个切片的重要性来自不同领域的关注。一个病人没有活检从上叶,在疾病最显著的计算机断层扫描,诊断只有联合化疗后自信的审查。在其他情况下co-existent纤维化在计算机断层扫描发现,活检没有从降低叶;因此,它仍不确定是否这些地区的延伸PPFE低叶或代表另一种疾病,如摘要/ IPF,服务员治疗和预后的差异。
应该强调IAFE不是特定于PPFE和放射治疗的结果可以看出,随着化疗和作为一个续集某些吸入的损伤。IAFE的发病机理仍然知之甚少,但显然这是一个途径肺损伤常见的各种疾病,包括PPFE。IAFE也重叠的特点与顶帽(28,29日]。然而,顶帽是在解剖学上局部的,没有扩展到其他叶,除了极少数情况下,形成大规模的病变,这些不应该进入PPFE的鉴别诊断,因为实质组件不是顶帽的成像特性。
病人治疗管理在本系列中高度变量和主要经验,反映出在PPFE患者缺乏经验。临床过程是逐步在这些患者中,与先前的研究发现整合,尽管在某些情况下,积极的治疗包括糖皮质激素和免疫抑制剂。
本系列案件的局限性是它的回顾性本质和事实数据没有完全完成,但我们相信,这一群人,迄今为止最大的西方文学中,证实了PPFE作为一种独特的临床病理的实体。鉴于我们严格的入选标准,在我们的“纯”队列数量小于它可能。可以说,我们的一个情况下,回想起来,仍被排除在外的人给ABPA的潜在的重叠,但我们选择保持在系列强调点关于潜在的临床联系。
总之,我们已经描述了十二PPFE患者,疾病更广泛的区域比先前报道的参与。间接数据表明复发感染发病机制可能有作用,可能与遗传易感性和自身免疫机制的贡献。事实,许多患者PPFE演示功能同时共存的间质性肺疾病的警告是临床医生应该考虑扩散形式的PPFE和其他fibrosing肺部疾病的鉴别诊断。
脚注
感兴趣的语句
没有宣布。
- 收到了2011年9月22日。
- 接受2011年11月30日。
- ©2012人队











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}