





文摘
支气管扩张是一种慢性肺部消耗性疾病,特点是不可逆转的扩张支气管的气道损伤和重塑的结果由于复发性或慢性气道炎症和感染。根本目的:包括自身免疫性疾病、严重感染、基因异常和后天失调。
支气管扩张的发病机制了解甚少。三个不同的致病的元素,即感染、炎症和酶的行动,相互作用,与支气管扩张的病理生理学。
一些最近的观察表明,气道炎症在支气管扩张来自一个管制细胞因子网络独立于气道细菌殖民化。
在目前的审查,目前了解细胞和分子炎症发生在宿主-病原体相互作用的动态过程,发挥相关作用气道壁的破坏导致支气管扩张的致病机制进行了讨论。
支气管扩张是一个渐进的和使人衰弱的疾病的病理描述航空公司成为永久性扩张的结果inflammatory-related破坏支气管壁的结构组件1,2。一般来说,如表1所示⇓支气管扩张,代表一些病理过程的最后阶段,从异物梗阻感染后损伤,遗传缺陷,改变宿主防御和自身免疫性疾病3。然而,根本原因可能无法识别在大约50%的情况下4。
这种疾病可以体现在两种形式:局限于一个领域(局部)或更广泛(普遍),常常伴随着鼻窦炎和分散的气流阻塞2。临床课程的特点是持续或复发性咳嗽、脓性痰生产,偶尔咯血,反复发作,疲劳和呼吸短促1。
虽然原发性支气管扩张患者早发性疾病,现在发病的年龄已经成为后来由于改善卫生设施,引入儿童免疫接种和早期和频繁使用抗生素。
疾病的临床表现是广泛的。一小部分病人,发病原因不明,发展早期和快速进行性疾病。另一个子集的病人,占据了绝大多数,似乎慢慢恶化几十年来从恶化频率的增加,痰量和支气管扩张的程度。最后,一些病人,通常那些单一叶参与,可以无症状发作与不恶化,即使几十年5。
的生活质量受影响的对象往往是严重受损,由于频繁的招生到医院,疾病的社会经济成本很高6。
发病的机制导致支气管扩张很复杂,到目前为止,不清楚。当前的观点认为夸张和不受控制的中性气道炎症反应引起下呼吸道细菌感染是致病的机制导致支气管扩张的第一步7- - - - - -9。基于这种观点,航空公司逐步受损是感染微生物之间的相互作用的结果,气道炎症和tissue-damaging物质分泌中性粒细胞9- - - - - -11。
一些观测表明,气道炎症在支气管扩张来自一个管制细胞因子网络12- - - - - -14细菌性呼吸道感染的,独立的。在囊性纤维化的早期阶段12,15- - - - - -17和支气管扩张14活跃的气道炎症已经报道,即使没有微生物感染。
气道炎症和感染
了解气道炎症的慢性气道炎症性疾病近年来大幅增加,由于应用程序的方法,如支气管肺泡灌洗(BAL)、支气管活检,最近,诱导痰18,19。然而,尽管相当多的理解正在取得的进展在哮喘气道炎症,慢性阻塞性肺疾病(COPD),所知甚少的支气管扩张20.。
宿主防御和炎症反应是基于一个复杂的细胞因子网络,导致细胞的激活和扩大参与免疫反应21。炎症反应的严重程度取决于促炎细胞因子之间的相互作用,调节,细胞因子和抗炎细胞因子和各种抑制剂,释放限制其范围和持续时间22。所以,促炎细胞因子释放反应损伤或感染时没有充分抵消了抗炎细胞因子导致局部或全身性病理21。
在原发性支气管扩张、慢性支气管感染和炎症相互作用负责进步的肺损伤23。
建立感染的动力学和随后的炎症反应的关系知之甚少24。一般来说,气道炎症被认为是呼吸道感染的结果,而不是它的前奏15。
异常细胞因子网络和/或不受控制的激活效应细胞可以体现为独立于炎症感染和/或炎症不成比例地增加或延长与细菌刺激的水平25。最近的证据表明,在支气管扩张,由细菌引发的气道炎症反应刺激过度与细菌的负担,甚至继续回荡在感染控制26。气道炎症反应的体内平衡改变细菌感染宿主-病原体相互作用的动态过程决定了肺部疾病的临床表现。
通常,支气管扩张气道是由细菌感染等流感嗜血杆菌,链球菌引起的肺炎和铜绿假单胞菌27,28。
铜绿假单胞菌感染
铜绿假单胞菌是一个投机取巧的病原体迅速清除从健康受试者的航空公司29日。囊肿性纤维化和支气管扩张学科它负责慢性感染23。
的毒性铜绿假单胞菌是多方面的,包括几个细胞相关,如弹性蛋白酶分泌蛋白质,磷脂酶C,和那些通过III型分泌系统内进行吗30.。
拉帕的e Silvaet al。31日表明,铜绿假单胞菌感染,通过细胞介导免疫反应,在支气管扩张的致病的机制有重要的作用。
肺泡巨噬细胞(AMs)和上皮细胞是一个主要的第一道防线铜绿假单胞菌肺部感染32,33。AMs刺激,分泌大量的促炎细胞因子和趋化因子,发挥直接作用在中性粒细胞的招聘26,34。
激活中性粒细胞,一些研究表明在囊性纤维化患者,已与突变的感应铜绿假单胞菌导致海藻酸生产和生物膜的形成35保护细菌免受激活中性粒细胞36,37。的黏液状的P。绿脓杆菌生物膜承受opsonisation和吞噬细胞的免疫系统38- - - - - -40论证一个增加公差,以及有毒的氧自由基35,41和抗生素42。由于这种突变,病人发展活力和持续的中性粒细胞炎症反应。虽然从nonmucoid黏液状的表型在囊性纤维化更相关,它也发现严重的支气管扩张和解决患者的气道阻塞的恶性循环,感染和炎症过剩,导致肺破坏和进一步损害清除的过程26。
有越来越多的证据涉及自然杀伤(NK)细胞铜绿假单胞菌肺部感染。NK细胞的表型差异所示鼠标菌株敏感和耐药慢性肺部感染铜绿假单胞菌43。
鸟型分支杆菌复杂的感染
鸟型分支杆菌复杂的环境中(MAC)无处不在,通常情况下,在健康受试者不致病的46,47。
MAC感染可引起支气管扩张和bronchiolectasis48,49在那些没有系统性免疫性疾病的迹象49- - - - - -51。这些病人是中年不吸烟的女性和特征计算层析发现周边结节和支气管扩张52,53。
病态,MAC肺部感染的特点是广泛肉芽肿影响航空公司造成气道狭窄和气道肌肉层的破坏,并可能导致支气管扩张49。
在MAC感染的特异性免疫反应,cd4阳性t细胞有重要的作用54,55。此外,NK细胞参与先天免疫反应对这种病原体55- - - - - -57。
一些观察,在体外和在活的有机体内表明,特定细胞因子及其相互作用可以提供保护或MAC感染的易感性。肿瘤坏死因子(TNF) -α和干扰素(IFN) -γ至关重要的发展对结核病和MAC保护性免疫小鼠46,54。老鼠缺乏IFN-γ或白介素(IL) -12年,IFN-γ的主要刺激生产,增加了敏感性结核分枝杆菌46,58,59和苹果46,60。
主题与遗传缺陷IFN-γ蛋白质或受体对分枝杆菌感染的易感性增加46,61年。据报道一些家庭MAC容易传播疾病,及其免疫功能低是由IFN-γ的产量下降造成的62年,63年或者通过IFN-γ受体异常64年。
流感嗜血杆菌感染
王et al。27有报道称,与支气管扩张和复发性感染nontypeable科目流感嗜血杆菌(NHTi)提供一个2型辅助细胞(Th2)主要反应生产的il - 4和il - 10。相反,在对照组中细胞因子模式与Th1反应是相一致的。因此,有人建议nonclearing适应性免疫反应下呼吸道的慢性感染支气管扩张科目有利于气道炎症过程27。
最近,据报道在活的有机体内NHTi形成附着生物膜的顶表面上气道上皮细胞65年。反过来反应通过增加上皮分泌的一些先天和适应性免疫因素调节气道炎症65年。流感嗜血杆菌刺激呼吸道上皮细胞巨噬细胞炎性蛋白的生产,引发和TNF-α在体外66年和在活的有机体内67年。
它也建议流感嗜血杆菌造成气道炎症可以作为网关的有机体,为殖民统治铺平了道路铜绿假单胞菌68年。
细胞在支气管扩张气道炎症的作用
中性粒细胞
中性粒细胞在先天免疫反应中发挥最相关的作用69年,70年。他们迅速迁移到炎症组织和提供强有力的效应机制如吞噬作用、生产活性氧介质和抗菌物质70年,71年。
中性粒细胞是主要的细胞存在于痰和BAL流体的支气管扩张72年,73年。在临床稳定的支气管扩张气道中性粒细胞增加,包括无菌支气管,但它们的数量进一步增加当航空公司与潜在致病性微生物的领地13。
在支气管活检、嗜中性粒细胞高密度已报告在支气管粘膜固有层74年,75年。
中性粒细胞招募到航空公司涉及大量的炎性介质,包括IL-1β,引发,TNF-α和白三烯(LT) B49,10,13,76年,77年。
Transepithelial迁移的中性粒细胞炎症血管内舱到网站的监管是一个多步骤的过程涉及一系列协调之间的相互作用表面粘附分子表达内皮细胞和白细胞78年- - - - - -83年。粘附分子调节这一过程的三个家庭:selectins;的整合蛋白CD11 / CD18;和免疫球蛋白super-family,包括细胞间粘附分子(ICAM) 1,血管粘附分子1 (VCAM)80年,83年- - - - - -85年和CD4786年。
这些粘附分子调节il - 1、TNF-α脂多糖84年,85年,87年和引发88年航空公司的,都是丰富的主题与支气管扩张8,9,72年,74年,89年。
郑等。85年有报道血清E-selectin, ICAM-1和VCAM-1在支气管扩张方面明显高于对照组。有趣的是,血清水平的粘附分子与功能参数和痰批量生产表明粘附分子取代相关序列的事件导致支气管扩张85年。
最近,已经积累的证据表明toll样受体(通常)调节器neutrophil-epithelial细胞相互作用78年。通常识别特定pathogen-associate表达的分子模式和大多数免疫细胞,包括嗜中性粒细胞和上皮细胞90年。通常遇到特定pathogen-associate结构时,触发一个信号级联,导致核转录因子的激活(NF) -κB和其他转录因子91年。
风险中性粒细胞的toll样受体激动剂原因引发的生产,L-selectin从细胞表面脱落,upregulation CD11b / CD18生产超氧化物,增加吞噬作用90年。
增加TLR2的mRNA水平一直在报道痰上层的支气管扩张的对象92年。
附着中性粒细胞最后迁移到航空公司的指导下中性粒细胞chemotractant等因素引发,LTB493年- - - - - -95年和TLR4受体激动剂96年。
当激活时,中性粒细胞分泌蛋白酶,包括中性粒细胞弹性蛋白酶(NE), catepsin G和proteinase-395年。
东北有蛋白水解作用和诱导细胞因子的释放,如白介素、引发和集落刺激因子97年,98年。它也是一个强大的segretagogue,诱导粘蛋白基因MUC5AC的表达通过代细胞内活性氧(ROS)99年。
在稳态支气管扩张,痰NE水平与中性粒细胞的百分比,促炎细胞因子(引发和TNF-α)和痰量24小时,这是一个疾病活动的标志8。
也在刺激(TNF-α和IL-1β)中性粒细胞释放基质金属蛋白酶(MMPs)One hundred.- - - - - -102年。水平的提高MMP-8和MMP-9已报告与支气管扩张BAL流体的主题103年,104年。MMP-8和支气管扩张气道MMP-9表达式关联显著与气道中性粒细胞数量而不是疾病肺功能和扩展105年。
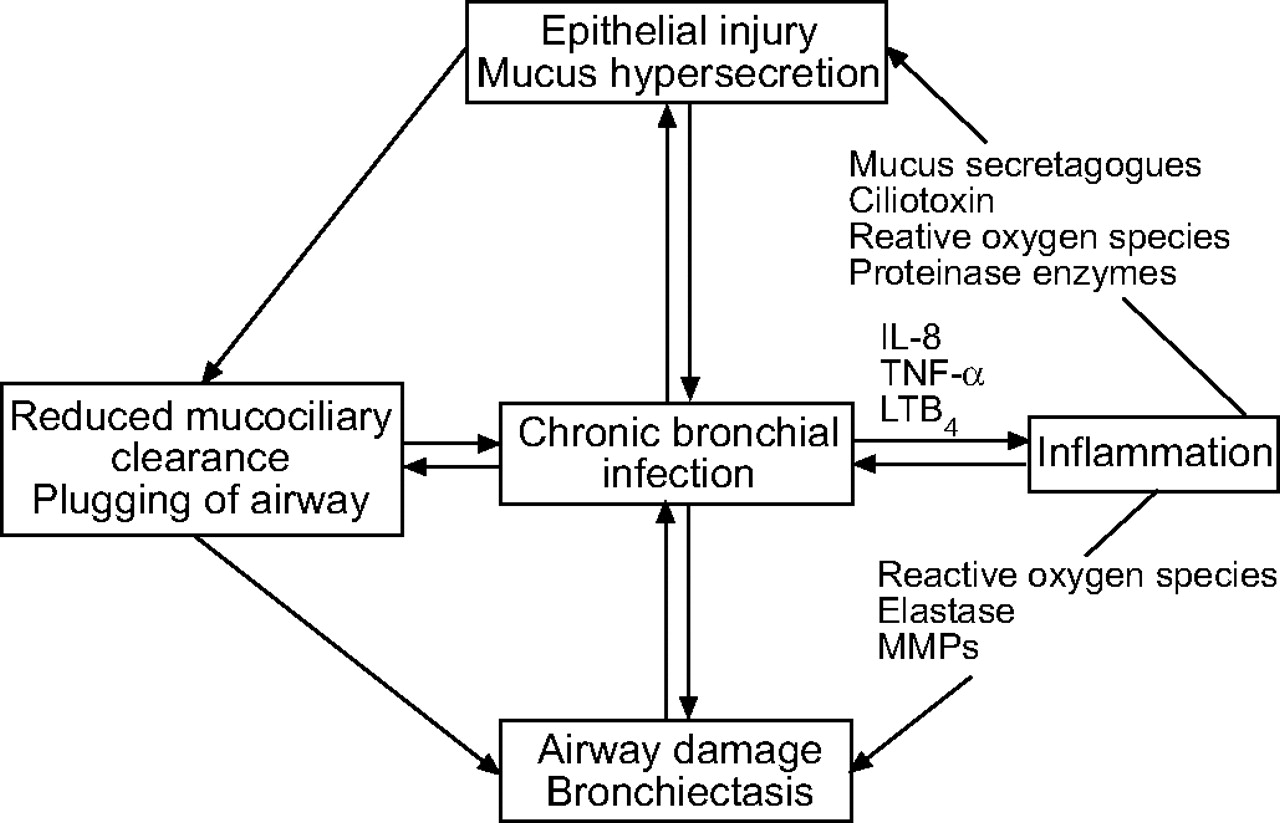
中性粒细胞的有毒产品损害呼吸道粘膜的结构和功能消化气道弹性蛋白,基底膜胶原和蛋白多糖,以这种方式造成病情恶化的疾病(图1所示⇓)8,99年,105年- - - - - -112年。
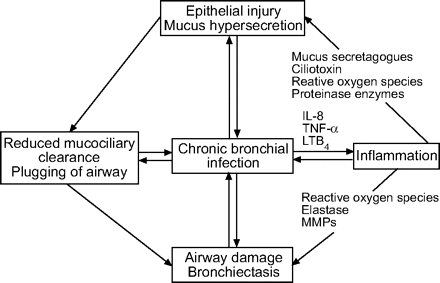
一个恶性循环的示意图表示的事件发生在慢性支气管感染。IL:白介素;肿瘤坏死因子:肿瘤坏死因子;LT:白三烯;MMP:基质金属蛋白酶。
巨噬细胞
巨噬细胞在支气管扩张的作用没有那么明确的中性粒细胞。支气管扩张的支气管活检,更高密度的巨噬细胞被发现在整个固有层与正常对照组相比74年。
更高数量的巨噬细胞在支气管扩张的主题报告与普通痰生产与nonproducers相比,表明气道巨噬细胞可能与疾病活动74年。
巨噬细胞促进中性粒细胞涌入到航空公司通过生产TNF-α75年和内皮素(ET) 1113年。他们也作为监管细胞释放多种炎症介质,包括TNF-α,引发,其他科学家趋化因子、单核细胞趋化现象的肽1,LTB4ROS和elastolytic酶的数量95年,114年,115年。
Vandivieret al。98年报道称,支气管扩张患者有更高水平的凋亡中性粒细胞比慢性支气管炎患者。
凋亡细胞为目标识别和吸收成phagocyte-expressing表面配体,特别是磷脂酰丝氨酸(PS),这与巨噬细胞表面的特异性受体98年,116年- - - - - -118年。
PS受体具有潜在的弹性蛋白酶乳沟网站,将导致损失的假定的PS-binding分子的地区98年,119年。据报道,不可以打通PS在吞噬细胞受体,影响凋亡细胞间隙,促成以这种方式进行支气管扩张气道炎症98年。
凋亡细胞的异常积累导致慢性组织损伤和炎症的持续98年,120年。事实上,炎症细胞死亡的凋亡是不可或缺的步骤在炎症反应的结束,和失败的这个过程可能会对航空公司的不利影响98年,121年。
淋巴细胞
在这两个实验模型31日和人类支气管扩张122年t淋巴球浸润航空公司。浸润t细胞观察,主要在固有层,但也分散在上皮。t细胞排列在上皮的基底膜,有时孤立但通常装在集群的细胞122年。
可用的研究结果在CD4 + / CD8 +比值产生了相互矛盾的结果。在一项研究中,CD4 +细胞普遍,CD4 + / CD8 +比值3:1的支气管扩张与对照组的1.3:1110年。在另一项研究BAL液体,CD4 +和CD8 +细胞的流式细胞术评估报告人数相等111年。
埃勒et al。72年发现CD8 +细胞主要在三分之二的患者在支气管活检从22日主题不同的病因学的支气管扩张。在剩下的活组织检查的部分中,CD4 +细胞的这些主要是集中在follicle-like结构72年。其他的研究已经报道的优势CD8 + t细胞31日,122年。然而,最近的研究表明,支气管扩张特征可能是通过增加CD4 +和CD8 + t细胞72年或者,事实上,没有优势的表型111年。
可用的数据表明,一个正在进行的细胞介导免疫反应导致的炎症过程中观察到支气管扩张31日,75年,122年。
细胞毒性t细胞的重要性是由一些观测对象与一种“光秃秃的淋巴细胞综合症”缺乏相关转运体(TAP)抗原呈递,不能把主要组织相容性复合体(MHC)类分子在表面。在这些患者中,CD8 + t细胞缺乏,因为他们不能积极在胸腺中选择,缺乏MHC类我表达。没有表达MHC类的我和CD8 + t细胞,这些病人遭受持续的呼吸道病毒和细菌感染。这些反复呼吸道感染的结果解剖损害的航空公司的发展,最终,导致支气管扩张123年- - - - - -125年。
NK细胞已经与家族性支气管扩张的发展主题,利用基因突变,损害类分子的表达,结果在NK细胞功能障碍123年。NK细胞在支气管扩张Boyton最近收到的支持工作et al。126年报道,主题与人类白细胞抗原(HLA) - c组1纯合性基因容易支气管扩张。HLA-c组1纯合性以及HLA-c /吉珥基因之间的相互作用导致过度或不适当的激活NK细胞,表明这些细胞的作用在疾病的发病的机制126年。
嗜酸性粒细胞
嗜酸性粒细胞的作用在支气管扩张的发病的机制,目前未知。支气管扩张,增加支气管粘膜的EG2 +嗜酸性粒细胞数量已经报道,尽管他们的数量小于中性粒细胞、巨噬细胞和t细胞数量75年。嗜酸性粒细胞数量的增加也被观察到支气管扩张患者的痰127年。
与特发性成年受试者支气管扩张、血清嗜酸性阳离子蛋白(ECP)水平已报告与性别和年龄相比显著增加控制和慢性阻塞性肺病患者。相比之下,血液嗜酸性粒细胞计数没有增加与正常对照组相比,或慢性阻塞性肺病患者128年。
气道嗜酸性粒细胞激活是慢性阻塞性肺病急性加重的特点和与疾病严重程度129年。
增加气道嗜酸性粒细胞浸润的临床和病理意义和支气管扩张的血清ECP水平科目,目前不得而知,但有可能是招聘的支气管扩张气道嗜酸性粒细胞和疾病严重程度相关。
上皮细胞
直到最近,呼吸道上皮细胞被认为是物理化学屏障所提供的几种本构先天机制(如。黏膜纤毛的清除、抗菌分子,常驻巨噬细胞和淋巴细胞),不断保护航空公司从有害吸入刺激物,病毒和细菌130年,131年。
有越来越多的证据表明,气道上皮细胞积极参与呼吸系统疾病的病理生理机制132年。支气管上皮细胞合成和释放大量炎性因子,持续和应对外界刺激133年,提供了一个当地的机制诱导,放大或调节持续的炎症132年。
人类支气管上皮细胞,当暴露在细菌内毒素,显著增加促炎症介质的表达和/或释放,包括引发和TNF-α,一个关键的角色在迁移和活化的中性粒细胞炎症的网站77年,133年。
人工气道上皮细胞产生ET-1,促进中性粒细胞粘附内皮细胞,移植到炎症和弹性蛋白酶的释放在体外113年。
支气管上皮细胞表达ICAM-1直接由上皮细胞与细菌和/或间接通过上皮细胞与其他细胞类型通信细胞因子134年。气道反应细菌病原体的调制是增加ICAM-1的表达式66年,134年。交互的ICAM-1 CD11 / CD18白细胞是白细胞粘附气道上皮细胞的行列式90年,135年和减少ICAM-1表达或功能可能导致白细胞招募受损和/或抗菌活性134年,136年,137年。
因此,目前的证据表明,气道上皮细胞是炎症反应的关键协调器通过释放促炎细胞因子和upregulation ICAM-1粘附分子。
上皮细胞表达通常,当激活,通过NF-κB和其他信号分子,导致转录upregulation炎症介质138年。通常也可以激活host-derived分子和膜脂质产生了组织损伤的结果139年。上皮细胞有许多相关的细胞表面分子的抗原,包括我和MHC II类和CD40 MHC类138年。在实验大鼠的支气管扩张模型已经发现上皮细胞表达MHC II级分子31日。感兴趣的是,出现二类MHC抗原在上皮细胞相关的组织学严重性bronchiectatic变化31日。
氧化应激
氧化应激指示由活性氧引起的。所有的功能和结构的变化140年- - - - - -142年。通常,毒性的氧化剂和保护性的抗氧化防御系统的平衡143年。呼吸爆发期间过度炎症细胞产生的大量自由基压倒宿主抗氧化防御系统造成严重损伤气道上皮细胞和其他气道结构144年,145年。然而,ROS能加重炎症,诱导细胞因子和趋化因子的生产通过刺激炎症基因由NF-κB控制146年。
氧化应激被认为发挥重要作用在支气管扩张和其他慢性炎症的病理生理学呼吸道疾病,如哮喘和慢性阻塞性肺病142年,147年,148年。
活化的中性粒细胞,嗜酸性粒细胞和巨噬细胞诱发呼吸道破裂导致生产超氧化物阴离子,然后经历了自发或此类歧化作用形成的过氧化氢(H2O2)144年。
水平的提高呼出H2O2在支气管扩张已报告主题140年。有趣的是,呼出H2O2水平与诱导痰中嗜中性粒细胞微分项显著相关,疾病,肺功能障碍和疾病严重程度73年。
中性粒细胞是一个内生的氧化剂来源73年,140年最近,它已经到了光TNF-α诱导中性粒细胞释放H2O2在体外149年。这意味着高浓度的细胞因子在支气管扩张可能会引发长期中性粒细胞释放H2O2140年。细菌感染可能导致氧化应激通过促进招聘和激活吞噬细胞的肺150年。事实上,支气管扩张的主题铜绿假单胞菌感染目前的水平明显高于H2O2相比之下,支气管扩张没有主题铜绿假单胞菌感染73年。
氧化剂损伤脂质(脂质过氧化)导致生产isoprostanes, 8-iso-prostaglandin F2(8-iso-PGF2)是best-characterised异构体。高浓度的诱导痰8-iso-PGF2已报告的主题与支气管扩张和哮喘151年,152年。
氧化应激指标的水平,如H2O2或8-isoprostane,呼出的气息凝结已被证明,以反映潜在的炎症的强度在支气管扩张140年、哮喘152年和慢性阻塞性肺病153年。
有各种各样的机制防止氧化应激。其中之一就是应激反应的诱导蛋白,血红素氧合酶(HO)142年,154年。HO-1,诱导HO,催化作用的血红素降解胆红素,产生自由铁和一氧化碳155年。包括图书馆在内的许多细胞因子和氧化剂TNF-αIFN-γH2O2和一氧化氮能够诱导HO-177年,154年,156年。的感应HO-1已被证明是参与解决急性炎症实验条件下,有人建议,它可能发挥cytoprotective作用在血红素和oxidant-induced细胞侮辱141年,157年。
水平的提高呼出一氧化碳在支气管扩张患者已报告反映增加氧化应激和顺向HO-1激活145年。
结论
在目前的审查,支气管扩张气道粘膜炎症事件的某些方面简要介绍。新兴的结论是,气管支气管扩张改变、独立的触发原因,来自于不受控制的招聘和激活大量的炎症细胞。每个炎性细胞的贡献和序贯作用涉及生产的典型病理支气管扩张仍知之甚少。
有数据表明支气管扩张气道炎症的结果从一个先天的放松管制,而且适应性,免疫反应在慢性呼吸道细菌感染。
可能不受控制的嗜中性粒细胞大量及其激活涌入航空是持久的结果激活先天免疫的效应机制。在不久的将来,还需要更多的研究来更好地了解先天免疫反应的信号机制,在先天免疫缺陷的本质与支气管扩张有关。细胞因子的能力精确控制炎症细胞的运动到红肿的航空公司表明,细胞因子及其受体可能提供目标治疗治疗调节气道炎症,为了防止进一步的肺功能恶化和更好地控制症状。
一些重要的问题仍未解决的,值得进一步研究。支气管扩张的假定触发事件比支气管扩张本身更常见。重要的是理解为什么一些人暴露于触发事件发展永久性损伤导致支气管扩张而其他人没有。同样,识别风险因素与疾病进展,尤其是儿童,尤其临床重要性因为这可以允许早期干预,减少长期的发病率和死亡率。
感兴趣的语句
没有宣布。
- 收到了2007年6月11日。
- 接受2007年9月24日。
- ©人期刊有限公司











{kind=link}
{kind=link}