





摘要
背景purinoceptor亚型P2X3.已经被证明与咳嗽反射有很大关系;purinoceptor的异质三聚体版本(P2X2/3)与味觉障碍有关。目前最先进的临床候选拮抗剂gefapixant对P2X的选择性较低3.而新开发的sivopixant对P2X具有较高的选择性3.与P2X2/3.
方法在一项2a期随机、双盲、安慰剂对照、交叉、多中心研究中,难治性或不明原因慢性咳嗽的成年患者接受口服sivopixant 150 mg或安慰剂,每日1次,持续2周,随后是2 - 3周的洗脱期,然后过渡到安慰剂或sivopixant,持续2周。评价sivopixant的疗效和安全性。
结果在31名随机分组的患者中,sivopixong -first组的15名和安慰剂-first组的15名完成了研究。治疗2周后,白天(主要终点)和超过24小时(次要终点)的平均小时咳嗽次数与基线的比值经安慰剂调整分别为- 31.6% (p=0.0546)和- 30.9% (p=0.0386)。Sivopixant还改善了与健康相关的生活质量。在sivopixant和安慰剂治疗期间,分别有12.9%和3.2%的患者发生了治疗相关不良事件。2例(6.5%)患者在给予sivopixant期间发生轻微味觉障碍。
结论Sivopixant降低了难治性或原因不明的慢性咳嗽患者的客观咳嗽频率,改善了与健康相关的生活质量,味觉障碍发生率低。
摘要
这项研究显示了高选择性P2X的疗效3.受体拮抗剂可减少咳嗽频率,味觉障碍发生率低。Sivopixant可能是难治性或原因不明的慢性咳嗽的一种有前途的治疗选择。https://bit.ly/3awojQH
简介
慢性咳嗽,定义为持续咳嗽8周,影响全世界约10%的普通人口,尽管有相当大的变异性(2-18%)[1,2].这种持续性和刺激性的情况可能导致生活质量受损和各种共病,包括大小便失禁、咳嗽性晕厥和肋骨骨折[3.].慢性咳嗽可能是由多种情况引起的,其中一小部分患者没有可确定的病因,称为不明原因慢性咳嗽(UCC) [4,5].大多数难治性慢性咳嗽或UCC (R/UCC)病例被认为具有咳嗽难治性的共同病理生理学,也称为咳嗽过敏综合征,由于外周和/或中枢敏感[6].目前,对于R/UCC患者,缺乏高效和可耐受的抗咳药;因此,有必要进一步研究开发治疗这种情况的药物。
purinoceptor亚型P2X3.是一个atp门控离子通道,主要表达在小直径初级传入纤维(Aδ和C)中,与感觉感知和传输有关。这种受体已被证明在咳嗽反射中有重要的作用。P2X有两种类型3.受体:同型三聚体(P2X3.)和异质三聚体(P2X2/3).P2X2/3受体与味觉障碍有关[7].Gefapixant,最先进的P2X3.受体拮抗剂在临床开发过程中,对P2X有3到8倍的选择性3.受体与P2X相比2/3受体(50%抑制浓度为30 nM的P2X3.P2X为100-250 nM2/3) [8].在一项临床试验中,gefapixant显著降低R/UCC患者的咳嗽频率,但以剂量依赖性的方式诱导味觉障碍[9,10].因此,一种对P2X具有高度选择性的化合物3.P2X亲和力低的受体2/3受体可能会有效地抑制咳嗽反射,味觉障碍的发生率较低。
Sivopixant (S-600918)是一种新开发的对P2X具有高选择性的化合物3.受体(与P2X2/3受体)在周围神经系统。通过构效关系研究,sivopixant被鉴定为具有高选择性的拮抗活性(50%的P2X抑制浓度为4.2 nM3.P2X为1100 nM2/3受体)和良好的药代动力学特性[11].
因此,我们开展了一项关于R/UCC的sivopixant概念证明研究,以评估sivopixant在R/UCC患者中应用2周的有效性和安全性。这项研究的一些结果以前已经以摘要的形式报道过[12- - - - - -15].
方法
研究设计和参与者
这是一项2a期,随机,双盲,安慰剂对照,交叉研究(图1)在日本的18个中心(包括10个专科诊所)进行了研究,包括年龄在20-75岁,R/UCC持续时间≥6个月的患者。使用视觉模拟评分(VAS)对患者进行咳嗽严重程度评估,评分≥40毫米,清醒≥10小时时的平均主观咳嗽频率−1在筛查期(1-4周)≥70%的天数(如患者日记所记录)前24小时内。这项研究得到了每个研究地点的机构审查委员会的核可,并按照《赫尔辛基宣言》进行。试验开始后,该方案未作任何更改。所有患者在入组前均给予书面知情同意。这项研究注册在日本制药信息中心临床试验信息数据库(japiccti - 184027)。患者资格标准,既往药物和有关研究方法的其他细节在补充材料.
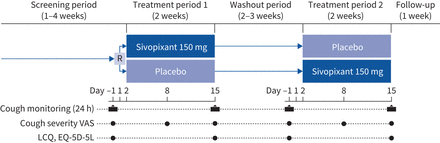
研究设计。第1天是−1的次日。R:第1天随机;VAS:视觉模拟量表;立法会:莱斯特咳嗽问卷(日文版);EQ-5D-5L: EuroQol问卷-5维度-5水平(日文版)。
治疗
入组患者按1:1分配比例被随机分配到sivopixantfirst组或placebo-first组。随机分组后,150 mg sivopixant(剂量基于一期研究结果;未发表的数据)或安慰剂,分别每天早上口服一次,持续2周。随后是2 - 3周的洗脱期,然后患者过渡到安慰剂或150 mg sivopixant 2周。第二疗程末次给药后随访观察7天。
结果测量
主要终点是sivopixant给药2周后,白天每小时平均咳嗽次数与基线的比值。次要疗效终点为:给药2周后24 h内、夜间、清醒和睡眠时每小时平均咳嗽次数与基线的比值;莱斯特咳嗽问卷修订(立法会;日文版(J-LCQ)) [16,17];VAS评估的平均咳嗽严重程度变化;及更改EuroQol问卷-5维度-5水平(EQ-5D-5L)(日文版)[18和EuroQol VAS (EQ-VAS)。
使用从VitaloJAK (Vitalograph, Buckingham, UK)咳嗽监测设备收集的数据来测量咳嗽的客观频率。对于J-LCQ,在LCQ总分中达到最小重要差异(MID)的患者比例(1.3分)[19评估。
安全性终点为不良事件(ae)和治疗相关ae的发生,以及其他安全性发现,包括血压、脉搏率、心电图和临床实验室检查。
统计分析
连续数据以均值和标准差表示,而分类数据以计数和频率表示。主要结果经安慰剂调整后,通过将混合效应模型应用于每个治疗期间sivopixant治疗2周后每小时平均咳嗽次数与基线咳嗽次数比值的公共对数作为响应来评估。主要疗效结果在全分析集(FAS)中进行评估。
在双面5%显著性水平下,保证80%的有效性所需的样本量计算为26。这是基于以下假设计算的:咳嗽的基线次数为30次,在接受sivopixant和安慰剂时,与基线相比的变化分别为−18和−3sd0.60的对数刻度。最终目标入学人数为30人。所有统计检验均采用SAS 9.4版本(SAS Institute, Cary, NC, USA)进行。
结果
病人
该研究于2018年8月20日启动,于2019年2月20日结束。本研究共纳入31例患者,随机分为sivopixant第一组(n=16)或安慰剂第一组(n=15) (图2).其中,sivopixong -first组和安慰剂-first组的15人完成了研究。FAS包括所有31例随机分组的患者。sivopixant-first组有1例患者因服用安慰剂期间出现紧张性头痛和体位性眩晕而退出研究。
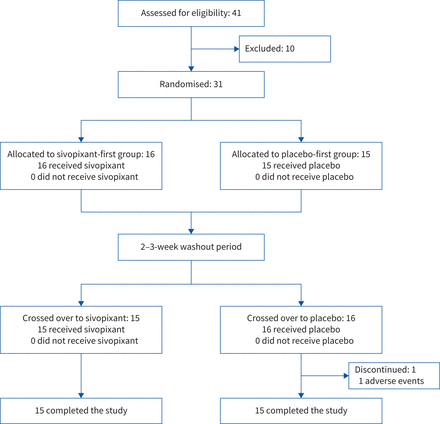
病人流程图。
女性占所有纳入患者的65% (表1).所有患者均为亚洲人。均值±sd患者年龄50.0±14.6岁,体重62.6±13.9 kg,体重指数24.1±5.0 kg·m−2.所有31例患者中最常见的基础疾病是哮喘(45%)、咳嗽变异性哮喘(32%)、鼻炎(19%)和胃食管反流疾病(16%)(一些患者有两种或两种以上的基础疾病)。19%的患者咳嗽原因不明。21例哮喘或咳嗽变异性哮喘患者既往曾接受过药物治疗,主要为吸入性皮质类固醇/长效β受体激动剂;视情况加入长效毒蕈碱拮抗剂或白三烯受体拮抗剂(补充表S1).均值±sd咳嗽持续时间为97.6±102.3个月。均值±sd1 s用力呼气量/用力肺活量比为82.1±7.5%。sivopixong -first组和安慰剂-first组之间的基线特征差异无统计学意义。基线咳嗽频率根据所接受的治疗报告在表2.
功效
对于主要疗效结果,研究药物给药2周后,白天每小时平均咳嗽次数与基线(主要终点)的比值为- 54.1%,而安慰剂为- 33.0%。经安慰剂调整后,sivopixant给药2周后日间每小时平均咳嗽频率与基线的比值为−31.6% (95% CI−53.6-0.8%;p = 0.0546) (表2).个别病人资料见图3.统计分析显示,未见顺序效应(p=0.8863)和周期效应(p=0.7238),且在第二个治疗期间,sivopixant组咳嗽次数减少。然而,在第二个治疗期间,在基线时,白天每小时咳嗽的平均次数(sivopixanfirst组为30.4次,安慰剂第一组为36.6次)低于第一个治疗期间的基线次数(sivopixanfirst组为53.4次,安慰剂第一组为59.1次),并且没有回到第一个治疗期间的基线水平(补充表S2).
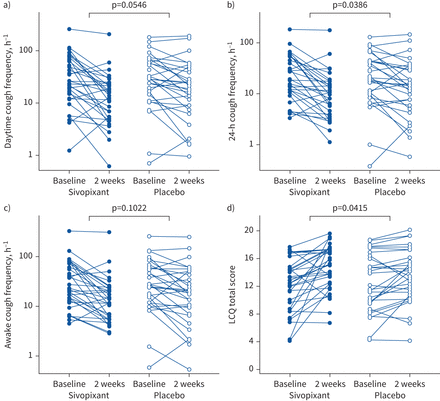
给药2周后24小时内每小时平均咳嗽次数与基线相比,sivopixant组为- 52.6%,安慰剂组为- 31.4% (表2).经安慰剂调整后,sivopixant给药2周后24小时内每小时平均咳嗽频率与基线的比值为−30.9% (95% CI−51.3 -−2.1%;p = 0.0386) (图3).至于夜间、清醒和睡眠时咳嗽,sivopixant给药2周后每小时平均咳嗽次数与基线的安慰剂调整比值低于安慰剂给药后,但差异无统计学意义(表2).
两周后,sivopixant和安慰剂给药后LCQ总评分与基线相比的变化分别为2.46和1.06,差异(sivopixant减去安慰剂)为1.40 (95% CI 0.06-2.75;p = 0.0415) (表3而且图3).sivopixant组LCQ总得分至少增加MID的患者比例为58.1% (95% CI 39.1-75.5%),而安慰剂组为35.5% (95% CI 19.2-54.6%)。2周后VAS显示,sivopixant给药后咳嗽严重程度的变化为−18.8 mm,安慰剂给药后为−12.4 mm(差异为-6.4 mm, 95% CI−14.8-2.0 mm;p = 0.1334)。
对于EQ-5D-5L评分,在给药2周后,sivopixant和安慰剂后,与基线相比的变化分别为0.10和0.01,差异(sivopixant减去安慰剂)为0.09 (95% CI 0.03-0.16;p = 0.0082)。对于EQ-VAS,在给药2周后,sivopixant和安慰剂组与基线相比的变化分别为11.4和2.8,差异(sivopixant减去安慰剂)为8.6 (95% CI 1.0-16.2;p = 0.0274)。
按方案集(PPS)分析包括24例患者:sivopixet第一组13例,安慰剂第一组11例。排除PPS的最常见原因是被禁止的伴随治疗(n=4)。此外,一名患者在测量过程中停止了咳嗽监测记录,另一名患者由于退出研究,在第二阶段没有咳嗽监测数据。
对于PPS,在服用研究药物2周后,白天每小时平均咳嗽次数与基线相比,sivopixant和安慰剂分别为- 54.8%和- 37.5%。经安慰剂调整后,sivopixant给药2周后白天平均每小时咳嗽频率与基线的比值为−27.7% (95% CI−52.6-10.4%;p = 0.1263)。
安全
安全性分析人群包括所有31例随机分组患者。在sivopixant治疗期间,35.5%(11 / 31)的患者发生了治疗紧急事件,而在安慰剂治疗期间,29.0%(9 / 31)的患者发生了治疗紧急事件。表4).1例患者出现两种治疗紧急事件(紧张性头痛和体位性眩晕),导致在安慰剂治疗期间停药。除与咳嗽监测装置胶带应用相关的接触性皮炎外,所有治疗紧急事件均发生在1 - 2名患者中(3名患者在使用sivopixant期间,1名患者在使用安慰剂期间;其中两名患者接受了局部皮质类固醇治疗,其他患者无需任何治疗即可自行康复)。
在sivopixant治疗期间,12.9%(31例中有4例)的患者发生了治疗相关的不良事件,而在安慰剂治疗期间,3.2%(31例中有1例)的患者发生了治疗相关的不良事件(表5).在sivopixant给药期间,各有1例患者发生了味觉障碍、味觉减退、口腔感觉减退和药物性肝损伤的治疗相关ae。1例患者在服用安慰剂期间发生口服感觉减退。没有死亡报告。然而,一名患者在服用安慰剂期间发生了严重的滑囊炎声发射(表4).
平均实验室值、血压和脉搏率的变化一般不大,在sivopixant或安慰剂治疗期间,随着时间的推移没有明显的临床趋势(数据未显示)。无临床显著心电图表现报告。
1例(3.2%)患者在给予sivopixant期间发生肝损伤。该患者有胆石症和肝脂肪变性病史,在研究期间接受熊去氧胆酸治疗胆石症。该患者的丙氨酸转氨酶和天门冬氨酸转氨酶水平在sivopixant给药第一周升高(虽然低于正常上限1.8倍),但在给药第二周下降到正常水平。继续治疗,患者在没有特殊治疗的情况下恢复。
讨论
这项交叉研究是第一个评估sivopixant的疗效和安全性的临床研究,sivopixant是一种高选择性P2X3.受体拮抗剂,在R/UCC患者。研究表明,sivopixant治疗2周(主要终点)后,白天每小时咳嗽次数较基线的变化大于安慰剂治疗后。然而,安慰剂调整后的差异没有统计学意义,主要终点没有达到。相比之下,sivopixant和安慰剂在24小时内每小时咳嗽次数上观察到有统计学意义,并且sivopixant改善了主观LCQ评分。很少有轻微的味觉障碍治疗-紧急的ae被观察到,没有患者因味觉障碍而停止。
sivopixant的安慰剂调整咳嗽频率比与其他P2X中报道的值相似3.受体拮抗剂试验。一项随机、双盲、对照、平行组2b期试验发现,每日两次gefapixant 7.5、20和50 mg导致清醒咳嗽频率的变化在- 22.0%之间(95% CI−41.8-4.6%;p = 0.097)和7.5毫克−37.0% (95% CI 53.3−−14.9%;P =0.0027)与安慰剂相比,剂量为50 mg [9].但是,由于P2X的作用3.味觉障碍是与P2X相关的最常报道的ae之一3.受体拮抗剂。年代mithet al。[9]表明,根据剂量的不同,10-81%的gefapixant治疗的患者报告了味觉相关的不良事件(味觉障碍、味觉减退或味觉衰老)。当gefapixant剂量为50mg(报告主要疗效结果有显著改善的最小剂量)时,48%的患者在治疗过程中出现味觉障碍,24%出现味觉减退,21%出现味觉减退[9].在研究中,在接受最高剂量治疗的患者中,与味觉相关的治疗紧急不良反应导致16%的患者停止治疗[20.].另一项随机、双盲、安慰剂对照、交叉、剂量递增的研究报告称,与安慰剂相比,gefapixant剂量≥30 mg对咳嗽频率的改善最大(p<0.05),也证实了味觉障碍以剂量依赖的方式发生[10].对于sivopixant,在本研究中报告味觉障碍的患者比例很低(2 / 31(6.5%))。此外,没有患者因味觉相关的治疗紧急事件而停止治疗。
较低的味觉相关治疗突发事件发生率是有利的,因为已知药物引起的味觉障碍会损害食物摄入,降低生活质量和治疗依从性[21,22].味觉障碍发生率低可能是由于药物对P2X的特异性3., P2X亲和性较低2/3.P2X特异性阻断3.由于P2X的作用更突出,因此预计不会导致味觉感知的显著损伤2/3在味觉感知和传递方面,P2X的比例较低3.受体与P2X相比2味蕾中的受体和参与味蕾反应转导的可能的通道冗余[7,23].最近对另一个P2X的研究也表明了这一点3.选择性拮抗剂(消光剂)[24].
在本研究中,sivopixant给药后24小时内每小时咳嗽次数与基线的比值显著低于安慰剂后的比值(p=0.0386)。然而,sivopixant治疗2周后,患者在白天、夜间清醒时和睡眠时每小时咳嗽次数的比值低于安慰剂治疗组,尽管存在差异与而安慰剂组则没有统计学意义。这些结果缺乏统计学上显著差异的一个原因可能是样本量不足。一般来说,慢性咳嗽在白天更为突出;因此,与咳嗽治疗相关的改善有望在白天更加显著,使其成为样本量计算的合理基础。正如预期的那样,在本研究中,安慰剂调整后的白天咳嗽的平均小时咳嗽频率减少比24小时咳嗽的减少要高(表2).然而,白天咳嗽的变异性比24小时咳嗽的变异性高,这可能会影响结果。我们的结果(图3)表明24小时咳嗽频率可能是药物疗效研究的最佳终点,使用白天/清醒咳嗽频率可能会错过治疗的临床疗效。
咳嗽频率的改善转化为主观测量的改善。在LCQ总分(p=0.0415)、EQ-5D-5L评分(p=0.0082)和EQ-VAS评分(p=0.0274)方面,sivopixant给药2周相关的安慰剂调整变化具有统计学意义。此外,与安慰剂组35.5%相比,sivopixant组58.1%的患者LCQ评分达到MID。一般来说,生活质量测量的显著改善不仅可以归因于咳嗽频率的改善,还可以归因于咳嗽强度的改善。虽然没有客观评估,但本研究可以假设咳嗽强度的改善。
在这项研究中,我们观察到,当患者在白天、超过24小时或清醒时服用安慰剂时,咳嗽次数减少了约30%。在之前的研究中也观察到类似的安慰剂效应,包括gefapixant的2b期研究[9], gefapixant三期研究中的安慰剂效应甚至更大[25].将目前的结果与以前的研究结果进行比较可能不合适,但不可能避免安慰剂效应,因为这是这类研究的一个特点。在这项研究中,安慰剂效应的原因尚未确定;然而,先前公布的gefapixant临床研究的阳性结果可能导致患者对慢性咳嗽的新疗法抱有很高的期望,从而促成了本研究中观察到的安慰剂效应。事实上,gefapixant的2b期研究报告中也提出了类似的建议[9].
这项研究的局限性之一是其交叉设计,其缺点包括一些潜在的遗留效应。洗脱期的长短是根据sivopixant的镇咳效果(不认为是由于任何有机变化)和单剂150mg sivopixant的半衰期来确定的,假设1周后血浆sivopixant浓度足够低。因此,我们认为洗脱期足以将任何遗留影响最小化。事实上,统计分析表明,没有观察到顺序效应和周期效应,这表明存在结转效应的可能性很低。其他限制包括纳入的患者数量少,只包括亚洲患者和一个国家(日本)。此外,咳嗽的原因和机制因人而异;因此,可以预期,不同的咳嗽病因对P2X的反应不同3.当前队列中的对立。在本研究中,45%的患者患有哮喘,32%的患者患有咳嗽变异性哮喘(合计77%)。哮喘相关咳嗽的发病率高于其他研究报告的发病率[26].与非哮喘性慢性咳嗽相比,哮喘相关咳嗽的特征是夜间咳嗽相对占优势[27- - - - - -29].与其他研究相比,这可能会降低白天咳嗽的相对发生率。此外,本试验中使用的剂量并不是基于临床疗效剂量范围研究的数据选择的。为了确定最佳的临床剂量,对R/UCC (ClinicalTrials.gov:NCT04110054).
总之,尽管由于未达到主要终点,因此应谨慎看待本二期研究的结果,但sivopixant在R/UCC中可有效减少咳嗽频率并改善与健康相关的生活质量,且很少有味觉干扰的不良事件。我们建议进行进一步的研究,如剂量发现研究和平行组3期试验,其中包括来自不同种族/民族和国家的更多患者。
补充材料
可共享的PDF
确认
感谢盐木奈健一(日本大阪盐野木株式会社)对本文撰写的宝贵支持。医疗写作支助由Ivan Olegario(新加坡MIMS私人有限公司)提供,由Shionogi有限公司资助。另外的写作协助由ProScribe - Envision制药集团(日本东京)提供,由盐野木株式会社资助。MIMS Pte Ltd和ProScribe的服务符合良好出版规范(GPP3)的国际准则。我们感谢Edanz (https://jp.edanz.com/ac)编辑部分稿件。
脚注
本文的补充资料可从www.qdcxjkg.com
本研究前瞻性注册于日本制药信息中心临床试验信息数据库的标识符为JapicCTI-184027。本研究的数据可根据医疗保健提供者、调查人员和研究人员的合理要求提供,以实现特定的科学或临床目标。盐野木株式会社致力于审查研究人员对临床试验方案、去识别的患者级临床试验数据和研究级临床试验数据的访问请求。更多信息请访问:www.shionogi.com/global/en/company/policies/clinical-trial-data-transparency-policy.html
作者贡献:a . Niimi, H. Ishihara, M. Machida和S. Miyazaki对研究的概念或设计做出了贡献。J. Saito, T. Kamei和M. Shinkai参与了数据收集。新井、斋藤、龟井、新海诚、石原、町田、宫崎等人撰写了论文的初稿。M. Machida进行了初步的统计分析。A. Niimi, J.斋藤,T.龟井,M.新海诚,H.石原,M.町田和S.宫崎在手稿开发过程中提供了书面意见和反馈,并直接参与了研究的执行。所有作者都认可了手稿的定稿。
利益冲突:A. Niimi没有什么可透露的。
利益冲突:J.斋藤没有什么可透露的。
利益冲突:龟井龟井没有什么可透露的。
利益冲突:新海诚先生没什么可透露的。
利益冲突:H. Ishihara是Shionogi & Co., Ltd的雇员和小股东。
利益冲突:M. Machida是Shionogi & Co., Ltd的雇员和小股东。
利益冲突:S. Miyazaki是Shionogi & Co., Ltd的雇员和小股东。
支持声明:本研究由盐野木株式会社赞助。主办方的科学代表参与了研究设计的开发。研究人员收集的数据由作者和主办方的代表进行分析。所有的作者,包括那些赞助商的代表,都可以完全访问研究中的所有数据。本文的资助信息已存入Crossref基金管理人登记处.
- 收到了2021年3月11日。
- 接受2021年10月5日。
- 版权所有©作者2022。
本版本按照创作共用署名非商业性许可4.0的条款发布。商业复制权利及许可请联系权限在}{ersnet.org











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}