





摘要
背景肺鳞状细胞癌(LUSC)在全球癌症死亡中占很大比例,其发病前气道上皮出现渐进性紊乱的侵袭性病变。然而,浸润前病变发展为浸润性LUSC的生物学机制尚不完全清楚。LRIG1(富含亮氨酸重复序列和免疫球蛋白样结构域1)在浸润前气道病变和浸润性LUSC肿瘤中下调,这与肺癌患者生存率降低有关。
方法和结果使用一个Lrig1在支气管镜下收集的敲入报告鼠和人气道上皮细胞,我们发现在稳态期间,LRIG1在气道上皮细胞中异质表达。在被怀疑是LUSC起源的基基气道上皮细胞中,LRIG1识别出具有较高的在体外小鼠和人类的增殖和自我更新潜能。利用n -亚硝基三氯乙基脲(NTCU)诱导的luc小鼠模型,我们发现Lrig1在侵袭前疾病的早期阶段,功能丧失导致异常高的细胞增殖,并形成明显较大的侵袭性肿瘤,表明疾病进展加快。
结论总之,我们的研究发现,LRIG1是一种具有高增殖潜能的基础气道祖细胞标记物,并且是浸润前肺癌进展的调节因子。这项工作强调了LRIG1的临床相关性,以及ntcu诱导的LUSC模型在候选肿瘤抑制因子和癌基因功能评估方面的潜力。
摘要
LRIG1在鳞状细胞肺癌的发展过程中丢失。本研究表明,LRIG1标记具有高增殖潜能的基底气道祖细胞,并调节浸润前鳞状细胞肺癌的进展。https://bit.ly/3AbPtY3
简介
肺癌是全世界癌症相关死亡的主要原因,每年诊断出210万例新病例[1].大多数患者患有晚期不治之症[2因此,制定早期发现和治疗策略是改善肺癌预后的关键。
85%的肺癌病例为非小细胞肺癌,其中三分之一为肺鳞状细胞癌[3.,4].LUSC发生于支气管上皮,先于浸润前病变的发展,从化生到越来越严重的不典型增生和癌原位(CIS) (5,6].侵袭性前鳞状气道病变与吸烟有关,吸烟是肺癌的主要危险因素[5,7,8].分子研究已经确定了侵袭前病变的遗传、表观遗传和转录变化[9- - - - - -13].然而,这些变化在肺癌发展中的生物学相关性尚不清楚。
富含亮氨酸重复序列和免疫球蛋白样结构域1 (LRIG1)是一种跨膜蛋白,作为表皮生长因子受体(EGFR)信号传导的负调控因子[14,15].LRIG1位于染色体3p14中,该区域常受侵袭前肺病变和非小细胞肺癌中拷贝数变化的影响[16,17].杂合子性的丧失LRIG1可见于75%的人类肺癌细胞系和低水平的LRIG1表达与非小细胞肺癌患者总生存率降低相关[18- - - - - -20.].我们之前已经证明,与供者匹配的健康上皮组织相比,侵入前CIS肺病变中LRIG1的转录物和蛋白质水平都较低[18],表明LRIG1在肺癌发生过程中具有早期作用。在这里,我们研究了LRIG1在正常气道上皮细胞中的表达,并检查了在LUSC发育过程中其功能丧失的后果。
材料与方法
老鼠饲养和实验
动物实验得到了伦敦大学学院生物服务审查委员会的批准,并遵照英国内政部的程序和道德准则进行。的C57BL / 6Lrig1: eGFP-IRES-CreERT2鼠系[21]两次回交FVB/N。小鼠饲养在C57BL/6和FVB/N混合背景的单独通风笼子中,昼夜循环12小时,并提供食物和水随意.将幼崽分成相应的实验组或对照组。
人体组织样本
伦理批准通过了国家研究伦理委员会(REC参考文献06/Q0505/12)。在自体荧光支气管镜检查中,从正常支气管上皮区域提取支气管样本。支气管刷直接用于流式细胞术和组织活检,冷冻在最佳切割温度(OCT)化合物(tissue - tek 4583)中。
流式细胞术
流式细胞术在Fortessa细胞分析仪(BD Biosciences)上进行,细胞分选在FACSaria (BD Biosciences)上进行。细胞周期分析用1 μg·mL培养活细胞−1Hoechst 33342在37°C下孵卵30分钟,然后进行抗体染色。所使用的试剂在补充表S1.数据分析使用FlowJo 10.0.6 (Tree Star)。
ntcu诱导的LUSC模型
取6周龄雌性小鼠背毛,取75 μL 0.013 M n -亚硝基三氯乙基脲(NTCU) (Santa Cruz sc-212265)稀释于丙酮,每周涂抹2次,连续12周。对照组只接受丙酮。对小鼠进行了11周的监测,并每周称重两次。献祭时,肺用4%多聚甲醛/PBS绝缘,4℃固定过夜,然后石蜡包埋。
组织学和免疫荧光
苏木精和伊红染色在自动染色系统(Tissue-Tek)上进行。采用标准方案进行免疫荧光和免疫组化染色。抗原提取方法、抗体、试剂和设备详细介绍补充的方法.
生物信息学分析
人肺细胞图谱单细胞RNA测序(scRNAseq)数据集分析[22]进行了详细的补充的方法.我们评估LRIG1从基因表达集合中下载的两组支气管镜下获得的人浸润前鳞状细胞肺癌病变的表达。第一个数据集GSE33479包含122个侵袭前病变样本,从正常上皮到侵袭性肿瘤,使用Agilent微阵列进行分析[10].第二个样本GSE94611和GSE108082包含来自51个CIS病变(进行性和退行性)的激光捕获上皮样本[11].数据在R统计环境(版本3.5.0;www.r-project.org)使用Bioconductor 3.7版本。
统计分析
使用Prism 7 (GraphPad)进行统计分析。测试和样本大小在图例中说明;P <0.05为差异有统计学意义。
结果
LRIG1在气道上皮细胞中异质表达
利用免疫荧光技术,我们已经证明LRIG1在小鼠上呼吸道中表达[18].为了表征其在气道上皮细胞不同亚群中的表达,我们使用了一种Lrig1敲入记者鼠标。在这个模型中,是盒式编码eGFP-IRES-CreERT2插入到下游Lrig1起始密码子(图1一个) [23].绿色荧光蛋白(eGFP)表达增强发生在内源性Lrig1启动子和导致功能缺失的等位基因。如前所述,皮肤和肠道[23],抗体染色证实eGFP在上呼吸道重复表达内源性LRIG1 (图1 b).
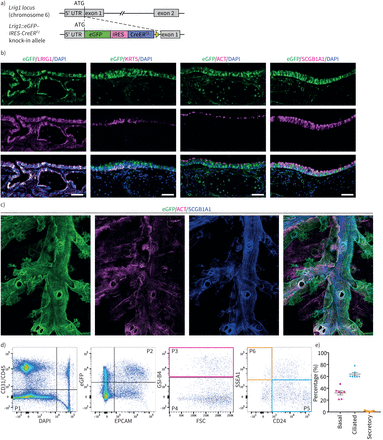
富亮氨酸重复序列和免疫球蛋白样结构域的表达1 (Lrig1)在老鼠的气道中。a)野生型和敲入报告器示意图Lrig1等位基因。增强型绿色荧光蛋白(eGFP)-内部核糖体进入位点(IRES)-CreERT2卡带的起始密码子是Lrig1将内源性基因移出框架。b)敲入报告基因杂合子小鼠气管切片的免疫荧光图像Lrig1等位基因表明eGFP(绿色)的表达再现了内源性LRIG1蛋白(洋红色)。eGFP的表达在气道上皮细胞的不同腔室中被检测到,包括KRT5+基底细胞,乙酰化微管蛋白(ACT+)纤毛细胞和SCGB1A1+分泌细胞。比例尺=50 μm。c)肺气道中表达eGFP的成年小鼠肺的全贴装制备。SCGB1A1和ACT抗体染色分别用于俱乐部细胞和纤毛细胞的鉴定。d)气管上皮细胞表达的免疫表型特征Lrig1: eGFP流式细胞术。活的气管上皮细胞(P1)通过表达CD31、CD45(内皮细胞和淋巴细胞)和4 ',6-二胺基-2-苯林多(DAPI)的细胞的负选择,然后是上皮细胞粘附分子(EPCAM)的正选择进行鉴定。+eGFP+细胞(P2)。Griffonia simplicifolia使用isolectin B4 (GSI-B4)标记来区分EPCAM内的基底细胞(P3)+eGFP+人口。分别通过表达CD24 (P5)和SSEA1 (P6)在gsi - b4阴性片段内选择纤毛细胞和分泌细胞。e) eGFP中细胞类型的分布+气管上皮(平均±扫描电镜, n = 7)。
在小鼠皮肤和肠上皮细胞中,LRIG1定位于确定的干细胞隔室,在那里它调节干细胞活性[24,25].相比之下,Lrig1: eGFP整个上气道上皮细胞表达明显。在基础(KRT5)中检测到eGFP+),纤毛(乙酰化微管蛋白(ACT+))和,在较小程度上,在俱乐部(SCGB1A1+)上皮细胞(图1 b).KRT5中未见eGFP的明显富集+粘膜下腺的细胞亚群,严重损伤后,粘膜下腺的干细胞库有助于表面上皮的再生[26,27) (图1 b).成年小鼠肺的全贴壁免疫染色显示Lrig1: eGFP表达扩展到整个支气管树,包括支气管和细支气管(图1 c).
研究基底细胞、纤毛细胞和分泌细胞在细胞群表达中的作用Lrig1: eGFP上气道上皮细胞采用流式细胞术。气管上皮细胞从携带一份Lrig1: eGFP记者等位基因。免疫和内皮细胞分别通过CD45和CD31的负选择被淘汰。Lrig1+上皮细胞粘附分子(EPCAM)和eGFP的双重表达鉴定上皮细胞。Griffonia simplicifolia隔离素B4 (GSI-B4),选择性地与气道基底细胞上发现的细胞表面碳水化合物结合[28,29],用于确定基底细胞种群。分别用CD24和SSEA1表达分离纤毛细胞和分泌细胞[30.) (图1 d).基底细胞构成平均值±扫描电镜的32.2±3.4%Lrig1: eGFP+纤毛细胞和分泌细胞分别占63.9±2.7%和1.3±0.3% (图1 e).选择最亮的eGFP后,细胞类型的分布没有显著变化+细胞(补充图S1),表明Lrig1基底细胞室的表达不丰富。只有50.8±4.1%的气道基底细胞人群Lrig1: eGFP+.
小鼠和人基础气道上皮细胞的表达Lrig1增加了在体外自我更新能力
只有气道基底上皮细胞亚群表达Lrig1,我们调查了其性质是否Lrig1-表达的基底细胞不同于Lrig1消极的程序。EPCAM+GSI-B4+从小鼠气管杂合子中分离出气道基底上皮细胞Lrig1: eGFP等位基因。细胞周期分析显示Lrig1: eGFP+基底细胞亚群中G2/M细胞的比例高于对照组Lrig1: eGFP- - - - - -分数(t检验p=0.0019) (图2一个这与之前的皮肤和胃的发现一致Lrig1表达识别更多增殖的干细胞/祖细胞群[21,23,24].

小鼠基础气道上皮细胞表达富亮氨酸重复序列和免疫球蛋白样结构域1 (LRIG1)增加在体外自我更新潜能。a)上皮细胞粘附分子(EPCAM)+Griffonia simplicifolia隔离素B4 (GSI-B4)+小鼠基础气道上皮细胞Lrig1: eGFP杂合子小鼠在CD31缺失后用流式细胞术从新鲜分离的组织中纯化+和CD45+人群。采用Hoechst 33342染色比较增强绿色荧光蛋白(eGFP)阳性和阴性部分的DNA含量。b) eGFP中G2/M期细胞比例较高+与eGFP相比- - - - - -基底细胞(平均±扫描电镜, t检验,n=7个生物重复)。c)单一eGFP+或eGFP- - - - - -用流式细胞仪将基底细胞分选到包含有丝分裂灭活的喂养细胞的96孔板的单个孔中。培养10天后评估产生含有>10细胞的菌落的细胞数量(配对t检验,n=4只小鼠)。d)和e) eGFP的能力+和eGFP- - - - - -在基质培养14天后,评估基底细胞形成球状体的情况(平均±0.01)扫描电镜,配对t检验,n=10只小鼠)。
以确定是否Lrig1-表达的基底细胞显示出更高的自我更新潜能,我们在二维培养中评估了其克隆潜能。刚分离的单株Lrig1: eGFP+而且Lrig1: eGFP- - - - - -来自报告等位基因杂合子小鼠的基底细胞被分类到96孔板,并在第10天评估集落形成能力。基底细胞表达Lrig1: eGFP形成的克隆明显多于没有表达的克隆Lrig1(t检验p=0.013) (图2 c).当被植入基质中时,基础气道上皮细胞会产生被称为“气管球”的三维类器官[31].在这种情况下,Lrig1: eGFP+基底细胞产生的气管球明显多于Lrig1: eGFP- - - - - -细胞(t检验p=0.002) (图2 de),表示Lrig1-表达的小鼠基底上皮细胞具有较高的在体外传播的潜力。
接下来,我们研究了LRIG1表达是否确定了人类气道上皮细胞中增殖更强的基底细胞群。查阅公开的人类气道上皮单细胞转录组资料(www.lungcellatlas.org) [32的研究结果表明,与老鼠相似,LRIG1由基底细胞、纤毛细胞和棒状细胞异质表达(补充图S2).使用来自人类肺细胞图谱的scRNAseq数据集[22,我们评估了LRIG1人支气管上皮中基底细胞不同簇内的表达。这揭示了LRIG1表达丰富为“增殖基底细胞”。“基底细胞”簇是静止的,在LRIG1负分数。在两个组分中均有“基部近端”特征的细胞,但在细胞中更为丰富LRIG1+一个(图3一b和补充图S3).在增殖的基底细胞群中,LRIG1水平与MKI67,它编码增殖标记Ki67(皮尔逊相关R=0.32 p=0.05) (图3 c).
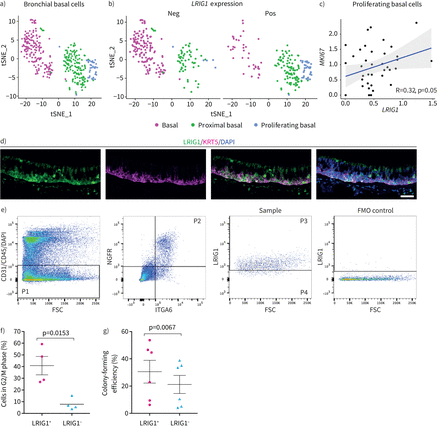
人基底气道上皮细胞中富含亮氨酸重复序列和免疫球蛋白样结构域1 (LRIG1)的表达与增殖增加有关。来自人类肺细胞图谱的单细胞RNA测序数据集被用于检查支气管基底细胞与支气管基底细胞之间的差异LRIG1表达式。a) t分布随机近邻嵌入(t-SNE)降维显示支气管区域主要基底细胞簇。b)含(Pos)或不含(Neg)的细胞计数LRIG1在每个基底细胞亚群中的表达。c)显示之间相关性的散点图LRIG1而且MKI67人肺细胞图谱中增殖基底细胞的表达。d)人气道上皮中LRIG1和基底细胞标记物KRT5的免疫荧光染色。比例尺=50 μm。e)整合素α-6 (ITGA6)+神经生长因子受体+用流式细胞术从支气管刷液中分离出人基底细胞P2,分离出LRIG1细胞+(P3)和LRIG1- - - - - -(P4)亚种。荧光减1 (FMO)控制显示。f)用Hoechst 33342对活细胞进行细胞周期分析,结果表明LRIG1+人基底细胞组分中G2/M细胞比例增加(平均±扫描电镜,配对t检验,n=4例)。g)单个LRIG1的比例+和LRIG1- - - - - -在有丝分裂失活的饲养细胞上培养10天后,人基底细胞产生集落显示(平均±扫描电镜,配对t检验,n=6例)。
LRIG1在KRT5中的表达+正常人支气管上皮基底细胞室经双免疫荧光证实(图3 d).用流式细胞术从自体荧光支气管镜检查中获得的支气管刷液中分离出正常的人基底细胞。去除CD45后+、CD31+气道基底细胞中富集的细胞表面蛋白整合素α-6 (ITGA6)和神经生长因子受体(NGFR)的共表达[31) (补充图S4),用来识别该细胞群。根据LRIG1免疫反应性(图3 e).活细胞周期分析显示,表达LRIG1的人基底细胞中G2/M细胞比例高于LRIG1阴性部分(配对t检验p=0.015) (图3 f).当评估菌落形成能力时,LRIG1+细胞产生的菌落比它们的LRIG1效率更高- - - - - -配对t检验p=0.0067 (图3 g).总之,我们的研究结果表明,基底细胞室中LRIG1的表达确定了一个增殖能力更强的细胞亚群在体外小鼠和人气道上皮细胞的繁殖潜力。
LRIG1是EGFR信号的负调控因子,因此LRIG1是EGFR信号的负调控因子+细胞表现出更大的增殖似乎是矛盾的。因此,我们假设LRIG1表达是祖基细胞增殖的一个检查点,但在低增殖人群(LRIG1阴性)中不需要。为了验证这一点,我们推倒了LRIG1在人类基底细胞中确实发现了增强的细胞群扩张补充图S5).因此,LRIG1标志着基底细胞的祖群,并作为增殖的检查点。
浸润前肺癌病变向浸润性LUSC的进展与减少有关LRIG1表达式
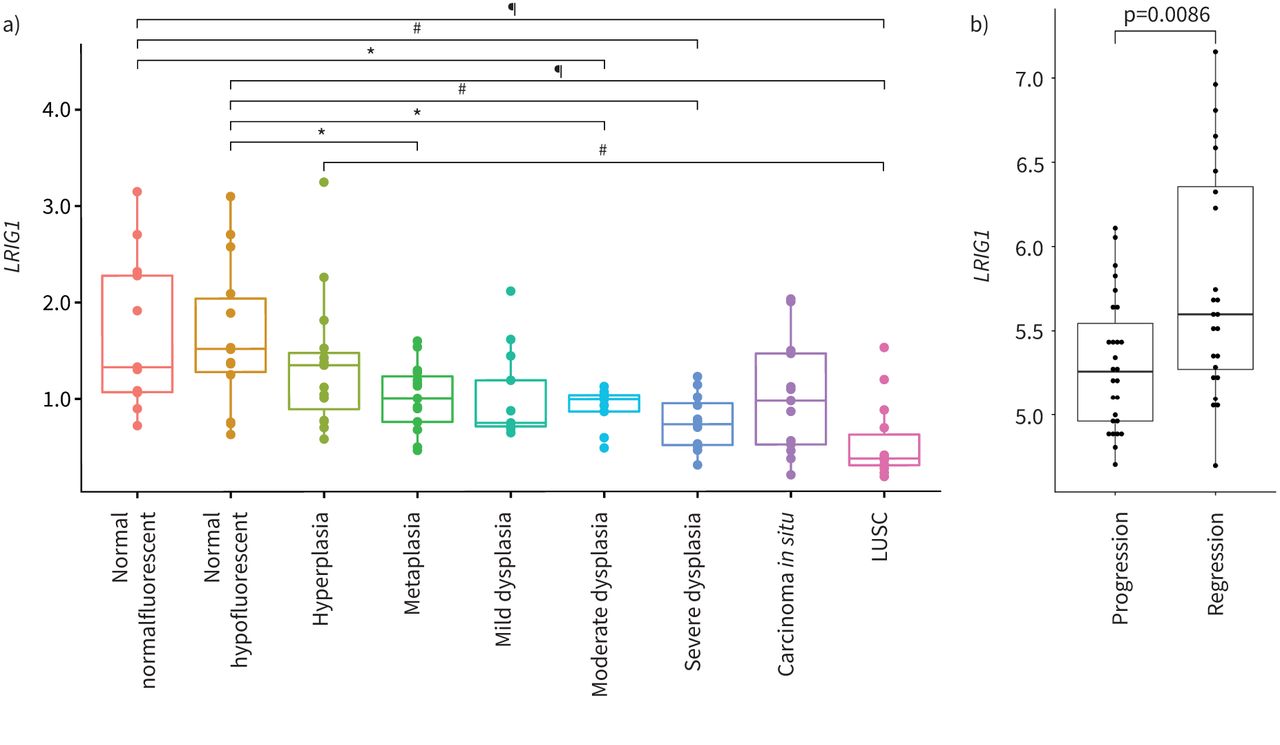
为了更好地理解LRIG1在LUSC形成中的作用,我们检测了LRIG1在人类luc发展的不同阶段。我们分析了已发表的数据集,包括来自正常支气管组织的患者活检,6种形态学上不同的侵袭前疾病等级,从增生到CIS和LUSC [10].这表明减少了LRIG1与正常组织相比,侵袭前样本中从化生到严重异型增生的表达不同(p<0.05) (图4一).进一步下降LRIG1与正常组织和增生性组织相比,观察到LUSC样品中的水平(p=2×10−5p=0.0026) (图4一).此外,评估LRIG1在进展为侵入性LUSC或退化的CIS病变纵向特征队列中的表达[11显示明显降低LRIG1进步组的水平(p=0.0086) (图4 b).总的来说,这些数据表明LRIG1表达与浸润前疾病到浸润性肺癌的进展严重程度相关。
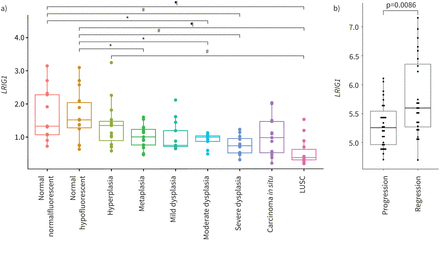
富亮氨酸重复序列和免疫球蛋白样结构域的表达1 (LRIG1)在人类肺鳞状细胞癌(LUSC)进展过程中。a)比较LRIG1根据标准组织学标准,在122个癌发展阶段的浸润前鳞状细胞肺癌病变中的基因表达。方差分析p = 3.7×10−7,采用Tukey多重比较检验进行组间分析。*: p < 0.05,#: p < 0.005,¶: p < 0.00005。b)分析LRIG1来自激光捕获癌的表达数据原位病变进展为癌症(n=27)或自发消退(n=24),显示表达减少LRIG1那些发展成癌症的人。p=0.0086 (Wilcoxon秩和检验)。
肺鳞癌小鼠模型的建立
调查减少的后果LRIG1我们使用了一个化学诱导的LUSC小鼠模型[33].将NTCU局部应用于年轻成年小鼠,每周两次,持续12周,结果在接下来的11周内luc形成(图5一个).组织学分析显示,ntcu诱导的病变再现了人类疾病的不同阶段,从侵袭前病变到侵袭性LUSC (图5 b).侵袭前病变和肿瘤表达luc标记KRT5和P63,缺乏肺腺癌(LUAD)标记表面活性蛋白C (SPC)的表达。罕见KRT5+在ntcu诱导的病变中检测到表达低水平SCGB1A1的细胞(图5 c和d)。
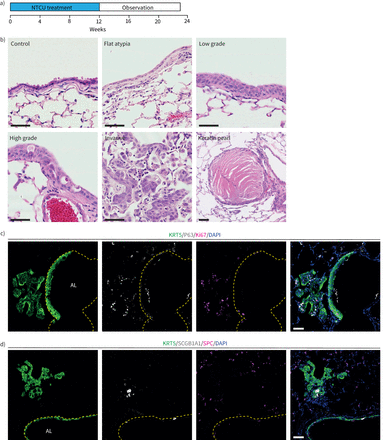
n -亚硝基三氯乙基脲(NTCU)诱导小鼠肺鳞状细胞癌。a) NTCU诱导小鼠肺鳞状细胞癌的给药方案。b)用苏木精和伊红染色的ntcu致肺病变代表性图像。扁平异型性,细胞核变大变平,核质比增加。低级别不典型增生,上皮细胞多层有序,从基底膜到管腔表面组织清晰。注意靠近腔腔的扁平核。高度不典型增生表现为上皮细胞层紊乱和多发核增大。浸润性鳞状细胞癌病变,开始充满肺泡间隙。角蛋白珍珠,鳞状细胞癌的特征性特征。c)免疫荧光染色显示肺鳞状细胞癌标记物P63和KRT5以及增殖标记物Ki67在ntcu诱导的病变中的表达。 d) Antibody staining demonstrates lack of immunoreactivity for the lung adenocarcinoma marker surfactant protein C within NTCU-induced lesions. Basement membrane (dashed lines) and airway lumen (AL) are indicated. Scale bars=50μm。
Lrig1在小鼠LUSC模型中,功能的丧失导致肿瘤大小的增加
的Lrig1: eGFP-IRES-CreERT2等位基因敲入导致Lrig1功能丧失(图6).来确定是否减少了Lrig1基因剂量对luc形成的影响,我们比较了NTCU在Lrig1-null, -杂合子和野生型小鼠。对动物的体重变化进行监测,从第15周开始,治疗组和对照组之间有显著差异(双向方差分析,p<0.05) (图6 b).

富亮氨酸重复序列和免疫球蛋白样结构域的影响1 (Lrig1) n -亚硝基三氯乙基脲(NTCU)的功能丧失诱发肺癌。a) LRIG1在野生型小脑组织裂解物上的免疫印迹,Lrig1杂合的,Lrig1零老鼠。注意蛋白质水平的变化是基因剂量依赖性的。α-微管蛋白显示为负载控制。b) ntcu处理动物和对照组动物的体重(平均值±扫描电镜,Lrig1+/+n = 11,Lrig1+ / -n = 9,Lrig1- / -n = 7)。双向方差分析:时间效应(p<0.0001)和时间×治疗相互作用(p<0.0001)。箭头表示所有治疗组与对照组之间有显著差异。Tukey's多重比较检验,p<0.05。c)经ntcu处理的小鼠横肺切片,用苏木素和伊红染色,并进行KRT5免疫组化处理,以确定侵袭前气道病变和侵袭性肿瘤。d)野生型(+/+)下气道上皮KRT5表达异常的比例,Lrig1杂合子(+/−)和Lrig1-null(−/−)小鼠NTCU治疗(平均±扫描电镜, Kruskal-Wallis检验,p=0.033,其次为Dunn多重比较检验,p=0.049;N = 7-11只/组)。e) krt5表达的病变分为扁平异型、低级别和高级别。下气道内不同等级浸润前病变的分布情况(平均±扫描电镜).双向方差分析:基因型效应(p=0.009)和病变等级效应(p<1×10)−6).平型异型的存在在基因型之间有统计学差异(Tukey's多重比较检验,p=7.7×10)−4, p = 1.7×10−5).f) KRT5染色的ntcu诱导的侵袭前病变代表性图像。比例尺=50 μm。g)每只小鼠的浸润性肿瘤数量。Kruskal-Wallis结果显示各组(每个基因型n= 7-11只)间无显著差异。h)不同小鼠浸润性肿瘤的大小Lrig1基因型(平均±扫描电镜).Kruskal-Wallis检验,p=0.0002,其次是Dunn多重比较检验,p=0.005, p=0.0003;Lrig1+/+n = 23日Lrig1+ / -n = 142,Lrig1- / -N =43个浸润性病变。包含IHC:免疫组织化学。
KRT5+基底细胞局限于小鼠上气道上皮[34].由此可见,KRT5在气管外及左右主支气管分支的表达是异常的。因此,我们使用KRT5免疫染色来评估NTCU治疗后支气管树中是否存在浸润前病变。计算每只小鼠浸润前病变总支气管树的比例(图6 c),并比较Lrig1-null, -杂合子和野生型小鼠。Lrig1基因型对侵袭前病变的程度有影响(Kruskal-Wallis检验,p=0.033)Lrig1零(Lrig1−/ -)组显示下支气管整体上皮表面KRT5表达异常Lrig1杂合的(Lrig1+ / -)小鼠(邓恩多重比较检验,p=0.049) (图6 d).浸润前病变区域根据严重程度分为扁平异型增生、低级别异型增生和高级别异型增生(表1而且图6 e和f).组间低级别和高级别病变的比例相当(图6 e).然而,侵袭前疾病的早期特征——扁平异型性的出现,在老年患者中明显较低Lrig1与野生型和Lrig1+ / -小鼠(双向方差分析后采用Tukey多重比较检验,p<0.001) (图6 e).
接下来,我们评估了三组患者的浸润性肿瘤。随着年龄的增加,luc的发病率有增加的趋势Lrig1基因剂量减少(χ 2检验为趋势,p=0.03) (表1).当我们比较不同动物之间肿瘤的频率和大小时Lrig1基因型,Lrig1基因剂量对每个个体的肿瘤数量没有显著影响(图6克).然而,与LRIG1表达活跃的小鼠相比,缺乏LRIG1导致的肿瘤明显更大(Kruskal-Wallis检验,p=0.0002) (图6 h).肿瘤较大,伴较低频率的扁平异型性Lrig1-无肺,说明丧失Lrig1促进和/或加速侵袭性疾病发展为侵袭性LUSC。
为了确定LRIG1是否通过调节细胞命运或促进增殖来调节LUSC进展,我们研究了P63和Ki67在不同病变光谱中的表达。KRT5+根据病变的严重程度和表达每种标志物的细胞比例(无论表达水平)对病变进行分类。整体比例P63+在ntcu诱导的肺病变中的细胞没有明显的影响Lrig1基因型或病变等级(双向方差分析,p>0.05) (图7).然而,当我们仅比较同级别病变时,我们发现扁平异型性病变中P63的比例明显更高+细胞Lrig1与野生型小鼠相比(Kruskal-Wallis检验和Dunn's多重比较检验,p=0.04)。Ki67分数的评估+细胞显示不同病变等级间存在差异(双向方差分析,病变等级效应,p=0.0007)。当我们评估相同级别病变的基因型影响时,我们发现与野生型小鼠相比,小鼠缺乏Lrig1Ki67的含量也有所增加+扁平异型性病变细胞(Kruskal-Wallis检验,Dunn's多重比较检验,p=0.04) (图7 b).这表明LRIG1活性在早期侵袭前疾病中通过限制祖细胞增殖来抑制LUSC的形成。
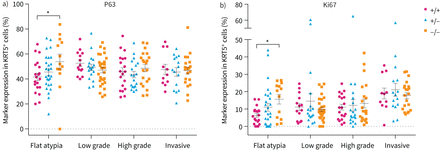
n -亚硝基三氯乙基脲(NTCU)诱导病变的特征。a)在野生型、富亮氨酸重复序列和免疫球蛋白样结构域中表达krt5的不同程度的气道鳞状病变中P63的表达1 (Lrig1杂合的,Lrig1-null小鼠(平均值±扫描电镜).b) krt5表达程度增高的鳞状气道病变Ki67免疫反应性Lrig1+/+,Lrig1+ / -而且Lrig1- / -老鼠(平均±扫描电镜).对于每个病变等级,影响Lrig1KRT5的基因型+对表达每种标记的细胞进行评估。Kruskal-Wallis检验,其次Dunn多重比较检验,*:p<0.05。对于a)和b) n= 11-31个病灶/分级/基因型;KRT5+细胞:Lrig1+/+n = 6776,Lrig1+ / -n = 206,Lrig1- / -n = 845。
讨论
对小鼠和人类内稳态上气道上皮细胞的研究突出了基底细胞室内表型和功能的异质性。目前的证据表明,基底细胞由多潜能干细胞和承诺祖细胞组成[32,35- - - - - -37].我们发现Lrig1在气道上皮细胞中是异质表达的,它的表达确定了基底细胞中增殖更强的亚群在体外自我更新潜能。这些发现与之前在表皮和前胃中的研究一致,在表皮和前胃中,LRIG1的表达标志着增殖性上皮干/祖细胞,在腺性胃上皮细胞中,LRIG1的表达标志着增殖性上皮干/祖细胞Lrig1-表达细胞的有机形成能力高于其Lrig1-负对应[21,23].这表明LRIG1在不同上皮细胞中调节祖细胞活性的保守作用。
相比之下,皮肤和肠道Lrig1在干细胞室中富集[24,25),在小鼠胃上皮中Lrig1由祖细胞和分化细胞共同表达[23,38].同样,我们发现在上呼吸道,表达Lrig1在基底细胞、纤毛细胞和分泌细胞中检测到。在小鼠远端气道,scRNAseq分析显示Lrig1在SCGB1A1中富集+中水平表达+SCGB1A1+支气管肺泡干细胞[39].对人类呼吸道的类似研究表明LRIG1在不同上皮细胞亚群中异质表达[32].这增加了LRIG1可能在气道上皮干细胞、谱系限制祖细胞和分化细胞中发挥功能作用的可能性。在未来的研究中,确定LRIG1失调如何影响这些不同人群将是很重要的,使用谱系特异性功能丧失或功能获得的方法。
LRIG1的失调在不同的肿瘤类型中可见[40].减少LRIG1在包括肺癌在内的一系列上皮肿瘤中,表达与不良预后相关[19,20.,41].我们现在展示的是LRIG1发生在LUSC进化的早期,在发展为高级别病变和浸润性肿瘤之前。原发性人类浸润前鳞状肺癌还没有在培养中培养,限制了在LUSC发展的这一阶段发生的分子改变的功能研究。我们发现LRIG1可拆卸的增强在体外从人类正常气道上皮细胞分离的原代基底细胞的繁殖,支持在侵袭性疾病发展中的潜在调节作用。最近,空气-液体界面培养的永生化人支气管上皮细胞丢失TP53而且SOX2过表达可重现支气管发育不良的特征[42].这一模型可用于评估损失的后果LRIG1在未来的研究中。
在ntcu诱导的小鼠luc模型中,我们发现luc的发展有增加的趋势Lrig1基因剂量减少。的缺失Lrig1导致扁平异型性的减少和形成更大的侵袭性肿瘤。在肝细胞癌中,LRIG1表达降低与肿瘤大小增大之间存在相关性[43].肺癌、乳腺癌和肝癌细胞系的功能获得和功能丧失研究表明,LRIG1调节癌细胞增殖[43- - - - - -45].评估Ki67在ntcu诱导的病变中的表达Lrig1在早期扁平异型性病变中,细胞耗竭导致异常高的增殖率,但在不典型增生或浸润性肿瘤中则没有。这些观察结果表明,较大的肿瘤Lrig1-无肺是疾病加速发展的结果,而不是肿瘤细胞增殖增加的结果。
我们已经证明,LRIG1在LUSC的发展中发挥肿瘤抑制作用。因此,LRIG1损失可作为侵袭前病变预后不良的生物标志物,导致较低的干预阈值。由于EGFR信号中LRIG1断裂的缺失导致了侵袭前疾病的发展,我们的工作强调了EGFR拮抗剂在LUSC预防中的潜在作用。其他数据表明,在抑制LUAD的关键作用。作为EGFR信号的负调控因子,LRIG1的高表达与LUAD患者2.8年的显著生存改善相关[20.].减少LRIG1LUAD细胞系,特别是EGFR突变的细胞系中的转录水平[44].转染egfr突变LUAD细胞LRIG1导致增殖、入侵和迁移潜力降低[44].
迄今为止,对LRIG1在NSCLC中的作用的功能分析依赖于在体外使用LUAD细胞系的研究和异种移植模型[18,44].在这里,我们使用了一种化学诱导的LUSC模型,该模型发展了内源性癌症病变,再现了人类疾病的组织病理学特征,包括侵袭前阶段的逐步进展。ntcu诱导肿瘤的RNA测序分析表明,它们的转录景观类似于人类的LUSC [46],支持该模型与理解LUSC进展的相关性。我们的研究表明,在转基因小鼠模型中使用ntcu诱导的癌变,可以评估LUSC发展的不同阶段的基因功能,并为验证候选肿瘤抑制因子、致癌基因和治疗靶点提供了一个系统,以预防和治疗LUSC。
补充材料
可共享的PDF
确认
我们要感谢Angela Barrett、Marie-Belle El Mdawar和Robert Hynds(伦敦大学学院,英国伦敦)分别协助进行定量分析、支持人类细胞培养和提供技术建议。
脚注
本文的补充资料可从www.qdcxjkg.com
作者贡献:L. Succony和S. Gómez-López进行了实验,分析了数据,并与S.M. Janes合作撰写了手稿。A. Pennycuick和A.S.N. Alhendi进行了生物信息学研究。D.戴维斯进行了细胞分类实验。S.E.克拉克和K.H.C.高尔斯对人类支气管上皮细胞的研究做出了贡献。N.A. Wright进行了病理评估。K.B.詹森提供了Lrig1: eGFP-IRES-CreERT2小鼠线和专家建议贯穿整个研究。L. Succony和S.M. Janes设计了这项研究。S.M. Janes监督了这项研究。所有作者都认可了手稿的最终版本。
利益冲突:L. Succony在研究过程中报告了来自维康信托基金会和英国癌症研究中心的资助。
利益冲突:S. Gómez-López在进行研究期间报告了来自英国皇家学会的拨款(牛顿国际奖学金)。
利益冲突:A. Pennycuick在研究期间报告了来自威康信托基金的资助(参考文献211161/Z/18/Z);其英国专利申请号为1819452.2。
利益冲突,A.S.N.阿亨迪没有什么可透露的。
利益冲突:D.戴维斯没有什么可透露的。
利益冲突:S.E. Clarke在研究期间报告了NIHR UCLH BRC的拨款。
利益冲突,k。h。c。高尔斯没有什么要透露的。
利益冲突:N.A. Wright报告来自英国癌症研究中心的资助,在提交的工作之外。
利益冲突:k·b·詹森没什么可透露的。
利益冲突:S.M. Janes在研究期间报告了惠康基金会的资助;Jansen和阿斯利康的顾问委员会工作的个人费用,GRAIL公司的拨款,在提交的工作之外。
支持声明:S.M. Janes是维康信托基金临床科学高级研究员(WT107963AIA),并得到了CRUK项目奖、Roy Castle肺癌基金会、Rosetrees信托基金、UCLH慈善基金会、Longfonds BREATH联盟和MRC UKRMP2细胞治疗平台的支持。Succony获得了CRUK临床研究奖学金和Wellcome Trust临床博士培训奖学金的支持。S. Gómez-López由英国皇家学会牛顿国际奖学金(NF161172)支持。Pennycuick是由维康信托临床博士培训奖学金资助的。这项工作部分由UCLH/UCL进行,他们从卫生部的NIHR生物医学研究中心的资助计划中获得了一部分资金。诺和诺德基金会干细胞生物学研究中心由诺和诺德基金会拨款NNF17CC0027852资助。本文的资助信息已存入Crossref基金管理人登记处.
- 收到了2020年3月23日。
- 接受2021年8月1日。
- 版权所有©作者2022。
此版本在知识共享授权许可4.0的条款下发布。











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}