





文摘
基本原理早产儿风险暴露于氧气在支气管肺的发育不良(桶),它的特点是肺增长逮捕。炎症是很重要的,但是机制仍然是难以捉摸的。在这里,我们调查了炎症途径和治疗目标严重临床与实验桶。
方法和结果首先,转录组分析在网上细胞反褶积了lung-intrinsic M1-like-driven细胞因子模式在新生鼠氧过多。这些发现证实了基因表达的macrophage-regulating趋化因子(Ccl2,Ccl7,Cxcl5)和标记(白细胞介素6,Il17A,Mmp12)。其次,hyperoxia-activated白介素6 (il - 6) /信号传感器和转录激活3 (STAT3)信号测量在活的有机体内和肺泡上皮细胞减少导致II型细胞(ATII)以及增加间充质标记。白细胞介素6空的老鼠表现出保存ATII生存,降低myofibroblasts和提高弹性纤维组装,从而使肺生长和保护肺功能。药物抑制全球il - 6信号和il - 6 trans-signalling提升alveolarisation氧过多后和ATII生存。第三,氧过多引发M1-like两极分化,可能通过4 Kruppel-like因素;hyperoxia-conditioned巨噬细胞IL-6-impaired ATII扩散媒介。最后,临床数据证明macrophage-related升高血浆细胞因子作为潜在的生物标记物,确定婴儿接受氧气的风险增加发展中桶。此外,macrophage-derived白细胞介素6和活跃的STAT3桶肺上皮细胞的相关损失。
结论我们提出一个新的IL-6-mediated机制在未成熟肺氧过多激活巨噬细胞,损害ATII体内平衡和破坏弹性纤维的形成,从而抑制肺增长。数据提供的证据表明,il - 6 trans-signalling可以提供一个创新药物目标使肺严重新生儿慢性肺部疾病的增长。
文摘
M1-like巨噬细胞活化与il - 6 / STAT3轴在临床和实验桶。macrophage-related抑制il - 6 trans-signalling促进肺ATII生存和生长在实验桶作为一种新的治疗早产儿。https://bit.ly/3AhF7GP
介绍
早产儿往往需要呼吸支持,包括机械通风和/或补充氧气。然而,这些拯救生命的治疗当应用于未成熟肺损害肺生长,导致新生儿慢性肺病,最初被描述为支气管肺的发育不良(桶)1]。肺的婴儿患有BPD通常的特点是减少肺泡的形成和摄动矩阵改造,产生结构性变化,青少年可能会持续超过(2,3]。尽管新生儿管理重大进展在过去的几十年中,桶的发病率居高不下,缺乏有效的预防和治疗策略。因此,识别新的分子通路损害肺泡生长和再生后未成熟肺的损伤可能导致潜在的新疗法。
肺泡上皮I型(ATI)和II型(ATII)肺泡内皮细胞暴露于氧过多时受伤,这可能导致桶的发病机制(4,5]。肺泡再生主要是由ATII,具有自我更新能力和产生ATI后肺损伤(6]。中断ATII体内平衡是参与各种慢性肺部疾病的发病机制,包括桶(7,8]。氧过多会降低生存和诱导上皮间充质转变ATII [9,10]。生长因子和细胞因子可以调节ATII体内平衡,减少肺泡形成和再生(10,11]。因此,肺氧过多后微环境的变化可能会复杂化的过程中桶通过减少ATII生存和肺泡的增长。
桶的发病机制还包括免疫细胞比例的变化,但不同的免疫细胞的机制和作用在桶发病机理不清楚(12,13]。最近的研究表明,居民企业和非居民巨噬细胞炎性肺的关键成分环境伤害,策划通过分泌多种细胞因子,包括白介素6 (il - 6)、细胞内稳态的调节器,矩阵改造,免疫反应和组织再生14]。另一方面,巨噬细胞也有助于再生领域,对于肺损伤后修复(15]。抗炎方法减弱alveolarisation受损引起的氧过多(16,17]。然而,氧气的影响的调节炎性肺微环境及其影响ATII体内平衡和肺泡形成仍然难以捉摸。
在这项研究中,我们提出的证据表明,长时间接触氧气概括严重新生儿慢性肺病、激活巨噬细胞,有利于M1-like两极分化,并增加il - 6在新生儿肺活动。值得注意的是,我们证明特定的封锁白介素trans-signalling保存ATII后生存和增长使肺氧过多。检查参与组成分泌腺的机械化,我们表明,hyperoxia-exposed的巨噬细胞抑制ATII生存,和我们确定一个可能的转录因子的小说角色Kruppel-like因子4 (Klf4)强调氧过多后巨噬细胞反应。在人类细胞培养的研究中,我们证明hyperoxia-activated炎性M1-like巨噬细胞有累加效应hyperoxia-reduced生存的人类肺泡上皮细胞(hAECs)。最后,我们确认macrophage-driven炎症il - 6 /信号传感器和转录的激活3 (STAT3)的血清反应存在于肺或婴儿用桶。
材料和方法
扩展的描述中提供的材料和方法补充材料。
动物实验
动物研究被分为四组:1)接触的男性和女性的新生鼠C57Bl6背景氧过多(80%啊2为14天)转录组的分析整个肺部,并40%或85%23、7或28天为后续评估的il - 6信号;2)接触新生儿C57BL6小鼠以及新生B6.129S2 - 3)白细胞介素6tm1Kopf / J(白细胞介素6−−/)和各自的野生型(WT)控制氧过多(85%啊2),28天来确定氧过多的长期影响。最后,4)新生鼠治疗腹腔内可溶性gp130Fc或在产后2天(P) il - 6马伯,P7,好,21岁。看到细节和描述的肺功能测试补充材料。
组织分析
信使RNA RNA提取,测量
RNA提取和定量实时PCR进行如前所述[18]。
细胞培养的研究
细胞系,试剂和化验
小鼠肺上皮细胞(MLE12;美式文化收藏)、巨噬细胞(J774A.1;美式文化集合),主要从WT小鼠,小鼠巨噬细胞(腹膜)人类monocyte-derived巨噬细胞从捐赠者和hAECs (CellBiologics;德国PELOBiotech)进行了研究。对于道德同意和详细信息,请参阅补充材料。MLE12和hAECs暴露在鼠标或人类il - 6 (Sigma-Aldrich、德国)+鼠标或人类IL-6Ra (IL-6Ra蛋白质、研发系统、美国)(il - 6 /可溶性白介素受体(sIL-6R))。扩散是评估使用MTT试验(写明ATCC;30 - 1010 k)。il - 6用ELISA测定(白介素ELISA、表达载体、美国)。Klf4沉默在巨噬细胞(J774.A1)和小干扰Klf4 (Dharmacon,猫。不。所以- 2589106 g,美国)使用Endo-Porter(美国Philomath GeneTools LLC)。
展示肺片(pcl)
小鼠肺孤立14-day-old小鼠气管内的安装后的琼脂糖。pcl治疗与il - 6 (Sigma-Aldrich、德国)+ 20 ng IL-6Ra (IL-6Ra蛋白质、研发系统、美国)(il - 6 / sIL-6R)下normoxia(21%啊2)或氧过多(85% O2)48 h。随后,pcl和4%多聚甲醛固定,石蜡包埋SFTPC和TUNEL检测免疫荧光染色(原位细胞死亡检测装备,罗氏公司、德国)。
人类的肺组织
桶和控制(十几)肺组织是通过国家心脏,肺和血液研究所核心Biorepository LungMAP财团人体组织。biorepository是罗切斯特大学研究对象审查委员会批准(RSRB00056775)。同意使用提供了每个样本的研究。临床元数据和组织病理学中列出表1。
人类的肺组织化学染色
桶和控制肺pSTAT3染色(1:200;细胞信号,没有。9145)、CD45 (1:200;英杰公司,14-9457-82),钙粘蛋白1 (1∶BD生物科学,610181)、CD68 (1∶eBioscience, 14-0688-82)和SFTPC (1:10 0, LSBio LS-B10952)。
原位杂交的人类肺
荧光原位杂交的白细胞介素6进行了使用先进细胞诊断RNAscope荧光多元分析。
临床研究
55个婴儿出生在< 32周妊娠被录取到一个纵向队列在西尔·帕卡德儿童医院的病人。斯坦福大学的机构审查委员会批准了这项研究。通知每个婴儿在父母的同意了这项研究。婴儿随访36周post-menstrual年龄。血浆样本化验为细胞因子分析63 -丛珠化验。临床资料总结补充表S2。
统计分析
连续测量数据都表示为平均值±标准误差的均值和p < 0.05表示显著差异。为在活的有机体内小鼠的研究中,双向方差分析或单向方差分析其次是邓恩的Bonferroni的测试后,分别Mann-Whitney检验和t检验是用来测试统计意义。细胞培养的研究中,我们应用(重复措施)单向方差分析后跟Bonferroni测试后或配对t检验。对人类的研究中,我们使用Mann-Whitney测试、t检验、非参数拟合曲线和混合线性模型。
结果
转录组分析确定趋化因子信号作为监管最严格的途径在小鼠肺氧过多
RNA转录组测序分析中所描绘的一样了图1一个。基因与平均log-fold变化大于2。错误发现rate-corrected p值< 0.01被认为是差异表达基因(度)(图1 bc)。KEGG通路数据库路径分析鉴定了大量的相关途径。其中,最著名的是cytokine-cytokine受体交互和chemokine-mediated信号通路(图1 d)。识别假定的功能网络,我们使用度的十大,衰减KEGG StringDB路径作为输入(图1情况)。首先,我们确定了10个最重要的生物过程(图1 e)和分子功能(图1 f使用基因本体分析)。其次,基于度的十大监管KEGG途径,我们生成的功能网络(图1 h),它强调的中心度。功能网络中的每个度高,它允许个人任务中所示的生物过程图1 e。我们下一个定义的中心度的排名,确定十大中心。这些中心,九是巨噬细胞趋化因子相关函数(图1 g)。
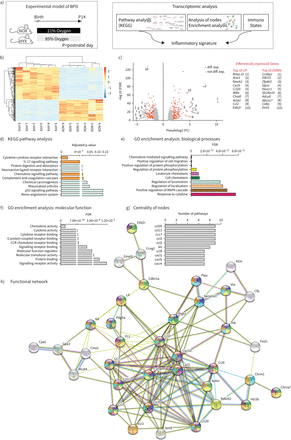
转录组分析肺的刚出生的老鼠暴露在normoxia(21%啊2、氮氧化物)或氧过多(80%啊2HYX) 14天(a),热图(b) (c)和火山阴谋。平均log-fold热图只包含基因改变(FC)大于2和错误发现率(罗斯福)—p值< 0.01。基因平均log-FC大于2和FDR-corrected p值< 0.01被认为是差异表达基因(diff. exp。),颜色为红色。标签在火山的情节显示前十——基于FC和抑制基因;每组n = 6;氮氧化物,normoxia (21% O2);HYX、氧过多(80% O2)。d)基因和基因组的京都百科全书(KEGG)路径分析显示肺的十大通路提供新生鼠暴露于氮氧化物或HYX 14天。超高频)功能富集分析识别功能基因集,包括生物过程(e)和分子功能的差异表达基因(f)使用确定的十大通路KEGG途径分析。度的排名(g)确认为节点的交互功能网络(h);n = 6 /组。桶:支气管肺的发育不良;Mmp10:矩阵metallopeptidase 10;Ano3:anoctamin 3;Nmrk2:烟酰胺核苷激酶2;Cxcl55:C-X-C主题趋化因子;Ccl20:20 CC-chemokine配体;IκBα:核因子k光多肽基因增强剂b细胞抑制剂,α;Chodl:chondrolectin;Acta1:肌动蛋白α1;Ccl2:CC-chemokine配体2;Eda2r:ectodysplasin A2受体;Crabp1:细胞视黄结合蛋白1;Glb1l3:牛乳糖beta 1喜欢3;Zfp663:锌指蛋白663;Glp1r:胰高血糖素肽1受体;Havcr1:甲肝病毒细胞受体1;Slc26a1026:溶质载体家庭成员10;Adcy8:腺苷酸环化酶8;Abca17:磷酸腺苷磁带,sub-family(他们),17个成员;CidecC:细胞死亡诱导DFFA喜欢效应;Vsnl1:visinin喜欢1;IL-17: interleukin-17;:基因本体;MAPK:增殖蛋白激酶;CCR:主题趋化因子受体;趋化因子受体Cxcr4:C-X-C主题趋化因子受体4。
在网上细胞反褶积标识炎性免疫细胞失调在新生儿肺氧过多
我们接下来immunoStates方法应用于基因表达数据来评估如果任何免疫细胞类型显示不同比例比normoxia氧过多。髓系树突状细胞、天真的B细胞和浆细胞增加;而CD4+、CD56+和肥大细胞被显著降低。与我们的假设一致,促炎M1-like巨噬细胞氧过多组的比例更高。相比之下,激活,抗炎M2-like巨噬细胞比例较低(图2一个b)。我们确认这些发现使用定量实时聚合酶链反应来确定关键的表达macrophage-regulating标记(Ccl7,Cxcl5和Ccl2)(图2 c)。此外,在肺氧过多增加额外的目标基因中观察到的新生鼠M1-like偏振编码白细胞介素6,Il17A和Mmp12(图2 d)。相比之下,M2-like标记,例如Il4,使用Il13和__arg1两组之间没有显著差异(图2 e)。评估基因表达和氧浓度的影响在早期发育的时间点,我们进行了定量实时PCR M1——M2-like标记在P3和P7在肺部的老鼠暴露到40%或85%2。基因表达分析显示明显lung-intrinsic增加白细胞介素6和Mmp12(M1-like标记),而M2-like标记(Il4和__arg1在P3和P7减少85%2(补充图S1a-d)。
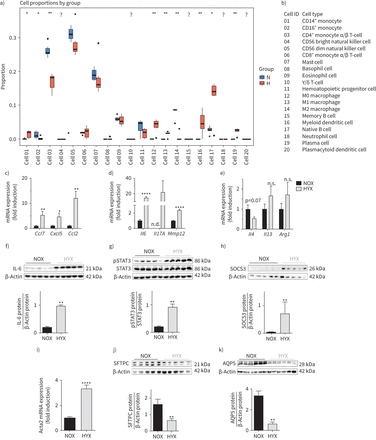
长期氧过多激活巨噬细胞和白细胞介素- 6 (il - 6) /信号传感器和转录激活3 (STAT3)信号在新生儿肺。免疫细胞类型)的比例在新生鼠肺暴露normoxia (N;21%啊2)或氧过多(H;80%啊2),14天。细胞比例被使用immunoStates估计在网上细胞反褶积每组(n = 6)。b)每个免疫细胞类型的识别号码列表。汉英)刚出生的老鼠暴露在normoxia(氮氧化物;21%啊2;黑条)或氧过多(HYX;85%啊2;灰色酒吧)28天,紧随其后的是评估基因表达的趋化因子确定为中央节点功能的转录组网络使用定量实时PCR和调节巨噬细胞功能。c) CC-chemokine配体7 (Ccl7),C-X-C主题趋化因子5 (Cxcl5),Ccl2。测量mRNA表达M1-like分化的基因特征。d)白介素6 (白细胞介素6),Il17A和金属蛋白酶12 (Mmp12)和M2-like分化。e)Il4,使用Il13和精氨酸酶1 (__arg1)使用定量实时聚合酶链反应(n =每组6 - 7)。f) il - 6蛋白丰度测量肺部免疫印迹的刚出生的老鼠暴露在normoxia(氮氧化物;21%啊2、黑条)或氧过多(HYX;85%啊2灰色酒吧)产后第一天(P1) P28每组(n = 5);β-actin担任加载控制;免疫印迹的光密度分析如下所示。g h)磷酸化和总STAT3 (pSTAT3 STAT3;g)以及SOCS3蛋白(h)评估免疫印迹;光密度总结数据显示相对于总STAT3和pSTAT3 SOCS3相对于β-actin,担任加载控制每组(n = 5)。我)测量α平滑肌肌动蛋白(αsma,被称为Acta2)mRNA在肺间质标记P28定量实时聚合酶链反应(n = 10 - 12 /组);基因表达是显示为折叠感应。j, k)免疫印迹显示上皮细胞标记P28:表面活性剂蛋白C (SFTPC)作为指标的肺泡上皮II型细胞(ATII) (j)和水通道蛋白5蛋白(AQP5)作为标记的ATI (k);β-actin担任加载控制;了各自的immunblot光密度分析如下所示。均值±扫描电镜。Mann-Whitney测试;*:p < 0.05;* *:p < 0.01;* * * *:p < 0.0001。
il - 6 / STAT3信号在新生鼠肺氧过多失去上皮细胞和间充质增加有关标记
自白细胞介素6信使rna几乎是20倍调节肺的刚出生的老鼠暴露在长期氧过多(85%啊2),我们评估肺il - 6 / STAT3信号出生后早期(P3, P7)和P28。il - 6的显著增加mRNA在P3和P7有关显著激活其下游效应器,STAT3和目标基因的表达Socs3在第七页。暴露在低浓度的氧(40%啊2)的磷酸化STAT3 (p = 0.0556)和一个重要的表达Socs3在P3 (补充图S2)。为了研究慢性肺损伤严重的氧气后,我们继续实验O为85%2。P28,我们发现5倍il - 6蛋白增加,显著激活其下游效应器,STAT3和更高的细胞因子的表达抑制信号后3 (SOCS3)氧过多normoxia(相比图2 f-h和补充图S3)。这些炎性相关研究结果几乎5倍增加αsma的表达式,也被称为Acta2平滑肌细胞表达的间叶细胞标记和myofibroblasts (图2我),较低的蛋白质丰富的SFTPC和水通道蛋白5 (AQP5),分别ATII和ATI标记(图2 jk)。
损失在新生鼠il - 6保护肺泡形成氧过多
如果丢失,我们下一个测试白细胞介素6将从hyperoxia-induced保护新生鼠肺损伤并允许肺泡形成。尽管氧过多简化WT小鼠肺泡,径向肺泡数的减少和增加意味着线性拦截,这些异常是减毒的白细胞介素6−−/老鼠(图3 a - c)。同样的,白细胞介素6缺乏保护新生鼠肺泡表面积的增加50%的单一肺泡中观察到WT老鼠,反映的能力开发一个更大的整体的气体交换面积(图3 d)。
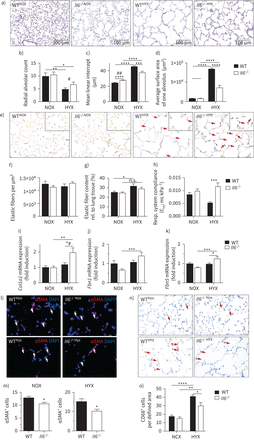
白介素6(损失白细胞介素6)保护肺泡弹性纤维的形成,并减少巨噬细胞入侵后在新生儿肺氧过多。)代表苏木精-和eosin-stained组织部分的图像显示在老鼠alveolarisation产后28天(P28)。野生型(WT)和白细胞介素6基因敲除小鼠(白细胞介素6−−/)暴露在氧过多(HYX;85%啊2)或normoxia(氮氧化物;21%啊2从产后第一天P28 (P1)。罪犯)汇总数据量化histomorphometric分析:径向肺泡数(b),意思是线性拦截(c),平均一个肺泡表面积(d)。e)代表哈特了染色组织切片的图像显示分布和弹性纤维在P28老鼠的本地化。箭头表示弹性纤维的二次氧过多后提示。做减法)汇总数据量化分析每个定义的区域(µm弹性纤维3)(f)和弹性纤维含量相对(rel)肺组织(%)(g) h)测量呼吸(职责)系统合规使用全身体积描记法白细胞介素6−−/并在P28 WT老鼠。i (k)基因的表达的评估肺矩阵的编码组件,包括弹性纤维。我)胶原蛋白1α1 (Col1a1)。j) Fibrillin 1 (Fbn1)。k) Fibulin5 (Fbln5)。l m)代表immunofluorescent染色α-smooth肌肉肌动蛋白(αSMA)作为myofibroblasts的标志。白色箭头描绘阳性染色细胞(l);αSMA的量化数据+细胞相对于所有4′,6-diamidino-2-phenylindole+(DAPI+)细胞(m)。n-o)代表图像使用CD68的巨噬细胞染色,×100放大。红色箭头描绘阳性染色细胞(n) CD68的量化数据+细胞每视野(o)意味着±扫描电镜;第4 - 9 n =每组。Mann-Whitney测试:#:p < 0.05;# #:p < 0.01。双向方差分析:*:p < 0.05;* *:p < 0.01;* * *:p < 0.001;* * * *:p < 0.0001;n。:not significant.
损失的il - 6保持弹性纤维组装和肺氧过多后合规
氧过多破坏弹性蛋白沉积在WT肺部,导致失去弹性蛋白二级septae和技巧的“我们”中的弹性纤维主要septae (图3 e)。相比之下,更少的肺组织弹性纤维检测hyperoxia-exposed白细胞介素6−−/老鼠,和本地化的二级septae出现类似于观察normoxia-exposed动物。绝对的每个定义的区域(µm弹性纤维含量2氧过多或缺il - 6)是不变的。然而氧过多,显著增加的相对弹性纤维含量但不是那样白细胞介素6−−/老鼠(图3 f在弹性纤维本地化,g)。这些变化可能占的保护白细胞介素6−−/从减少肺合规后氧过多(图3 h)。接下来我们分析,发现增加弹性纤维组件Col1a1,Fbn1和Fbln5信使rna在白细胞介素6−−/肺与WT (图3我- k)。此外,缺乏白细胞介素6与减少myofibroblasts normoxia和氧过多(图3 l, m)。尽管氧过多诱导巨噬细胞在WT和流入伊尔6−−/小鼠巨噬细胞丰度显著低于hyperoxia-exposed伊尔6−−/老鼠比WT (图3 no)。
il - 6促进表面活性剂损失蛋白表达和保护ATII氧过多
氧过多减少Sftpa和Sftpc信使rna在WT老鼠近50%,完全封锁的产生影响白细胞介素6−−/老鼠(图4一b)。根据这一发现,蛋白质丰富的SFTPC hyperoxia-exposed更高白细胞介素6−−/老鼠相比WT (图4 c)。使用SFTPC immunofluorescent染色,我们表明,氧过多SFTPC的数量减少+在两组细胞;然而,SFTPC+细胞在hyperoxia-exposed更高白细胞介素6−−/相比WT (图4 de)。最后,用TUNEL染色发现,细胞死亡,包括ATII hyperoxia-exposed低大约50%白细胞介素6−−/老鼠相比WT (图4 f- h)。
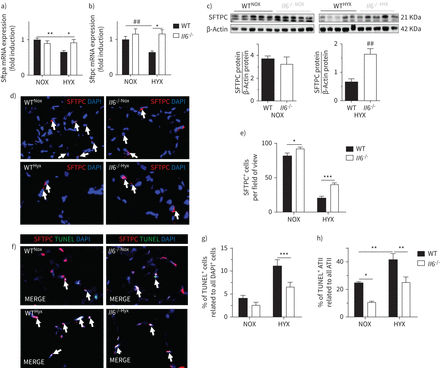
白介素6(损失白细胞介素6)保护肺泡上皮II型细胞(ATII)在新生鼠肺在暴露于长期氧过多。野生型(WT)和白细胞介素6基因敲除小鼠(白细胞介素6−−/)暴露在氧过多(HYX;85%啊2)或normoxia(氮氧化物;21%啊2从产后第一天P28 (P1)。a - b)评估基因编码的表面活性剂蛋白的表达(SftpP28)肺:Sftpa(一)和Sftpc(b)。c)免疫印迹显示表面活性剂蛋白c (SFTPC)蛋白质丰度在P28肺;β-actin SFTPC蛋白质有关,作为加载控制;密度测量数据显示低于相应的免疫印迹。表面活性剂蛋白质C d)代表免疫荧光染色(SFTPC;红色)作为一个指示器的ATII P28肺;4′,6-diamidino-2-phenylindole (DAPI)是用于核染色×100放大。SFTPC+细胞与白色的箭头表示。总结SFTPC e)定量数据+细胞每视野。f-h)代表co-immunofluorescence染色末端转移酶的dUTP尼克结束标签(TUNEL)作为细胞死亡的指标(绿色)和SFTPC(红色)在肺P28;DAPI用于核染色放大(f)×100。定量的百分比TUNEL的汇总数据+细胞(g)和TUNEL+相对于所有ATII ATII (h),意味着±扫描电镜;第4 - 9 n =每组;Mann-Whitney测试:# #:p < 0.01或显示;双向方差分析之后,邓恩的文章测试:*:p < 0.05;* *:p < 0.01;* * *:p < 0.001。
药物抑制全球il - 6信号和il - 6 trans-signalling
我们每周下对新生鼠,而暴露于氧过多,通过注射il - 6的马伯sgp130Fc,或者各自的车辆22)(图5一个)。虽然全球IL-6mAb中和il - 6, sgp130Fc只有块形成的复杂的il - 6和sIL-6R,叫做白介素trans-signalling。氧过多减少新生鼠的生存∼20%,但治疗与il - 6马伯或sgp130Fc保护老鼠(图5 b)。il - 6马伯和sgp130Fc保存alveolarisation肺部暴露于长期氧过多(图5 c- f),表明il - 6 trans-signalling通过sIL-6R部分底层机制。此外,我们发现,老鼠接受il - 6马伯或sgp130Fc氧过多ATII免受损失,相比有同样数量的ATII vehicle-treated老鼠normoxia (图5克,h)。
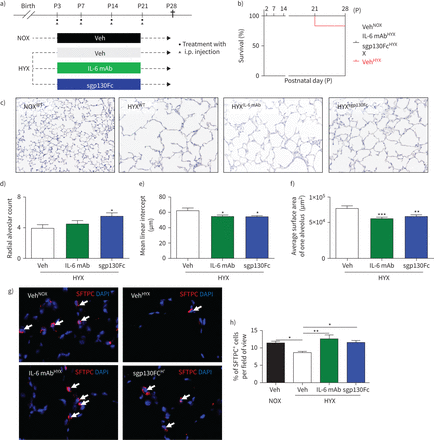
全球白介素6 (il - 6)信号和抑制il - 6 trans-signalling使肺生长促进ATII存活在新生鼠肺暴露于长期氧过多(HYX)。一)实验设计的方案。野生型(WT)小鼠暴露于氧过多(85%啊2从产后第一天(P1) P28;小鼠腹腔内(i.p)与车辆(阿明费)、il - 6马伯或sgp130Fc。b)存活曲线从出生直到P28: vehicle-treated老鼠normoxia (NOX);在HYX vehicle-treated老鼠;il - 6 mAb-treated HYX老鼠;并在HYX sgp130Fc-treated老鼠。苏木精- c)代表图像和eosin-stained组织部分显示alveolarisation P28在老鼠身上。d-f)定量histomorphometric分析汇总数据:径向肺泡数(d),意思是线性拦截(e),平均一个肺泡表面积(f)。为表面活性剂蛋白C (g)代表免疫荧光染色SFTPC;红色)作为一个指示器的II型肺泡上皮细胞(ATII)在肺P28;4′,6-diamidino-2-phenylindole (DAPI)是用于核染色×100放大。SFTPC+细胞与白色的箭头表示。总结SFTPC h)定量数据+细胞每视野%。均值±扫描电镜;n =每组4 - 6。单向方差分析Bonferroni紧随其后的测试后:*:p < 0.05;* *:p < 0.01;* * *:p < 0.001。
氧过多激活巨噬细胞通过Klf4和hyperoxia-exposed巨噬细胞的损失减少ATII的细胞生存
氧过多降低上皮细胞的基因表达标记Sftpc,但不Aqp5肺上皮细胞(MLE12)作为指标ATII ATI,分别。治疗MLE12与il - 6 / sIL-6R添加剂影响减少Sftpc,但不Aqp5(图6)。检查参与组成分泌腺,以确定是否对巨噬细胞(J774A1) MLE12调节细胞生存,我们暴露MLE12细胞氧过多,接着用标准单元中(Co), normoxia-conditioned巨噬细胞中(CM氮氧化合物)或hyperoxia-conditioned巨噬细胞中(CMHYX)。当CM的效果氮氧化合物类似于公司,厘米HYX减少MLE12扩散(图6 b)。我们下一个发现暴露巨噬细胞(J774A.1)氧过多24和48 h在体外显著增加il - 6蛋白条件培养基(图6 c)。此外,我们研究了氧过多激活M1-like分化的可能机制。之前的报告表明,失去Klf4促进M1-like分化(23]。的确,氧过多减少Klf4蛋白质培养巨噬细胞50%以上(图6 d)。此外,沉默的Klf4巨噬细胞强调的表达促炎症(M1-like)氧过多引起的细胞因子(图6 e)。我们的下一个主要暴露小鼠巨噬细胞(腹膜)WT小鼠氧过多,发现的表达显著增高Mmp12和Tlr4(M1-like)和显著减少__arg1和Fizz1(M2-like) (图6 f)。此外,厘米HYX进一步减少细胞增殖MLE12下氧过多相比,厘米氮氧化合物(图6克)。最后,我们暴露了pcl 14-day-old小鼠il - 6 / sIL-6R和normoxia或氧过多。il - 6在pcl引起细胞凋亡,SFTPC的重要添加剂对hyperoxia-induced细胞凋亡的影响+细胞(ATII) (图6 h)。
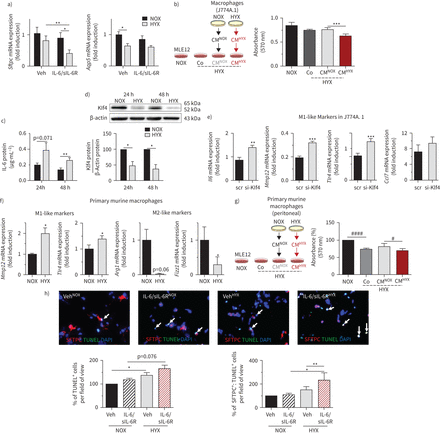
Hyperoxia-conditioned小鼠巨噬细胞和白介素6 (il - 6) /可溶性白介素受体(sIL-6R)触发hyperoxia-induced的肺泡上皮细胞凋亡II型细胞(ATII)。表面活性剂)评估基因表达蛋白C (Sftpc)和水通道蛋白5 (Aqp5)在小鼠肺上皮细胞(MLE12)。MLE12在serum-rich培养基培养,其次是serum-reduced媒介12 h。之后的细胞治疗与车辆(阿明费)或il - 6 / sIL-6R和暴露于氧过多(HYX;85%啊2)或normoxia(氮氧化物;21%啊2为24小时);n =每组11;β-actin担任管家基因。b)实验设计方案描述:巨噬细胞(J774.A1)暴露在氮氧化物或HYX 48 h;随后,MLE12受到HYX和处理条件培养液(CM) NOX-exposed巨噬细胞(厘米氮氧化合物),厘米HYXVeh(未经处理的介质,Co)或24小时;控制MLE12暴露于氮氧化物。最后,扩散MLE12使用MTT分析评估;n =夫人每组。c)测定il - 6蛋白使用ELISA J744上层清液。A1巨噬细胞。J744。一个1were cultured in serum-rich medium, followed by exposure to HYX or NOX for 24 h and 48 h; n=8 per group. d) Immunoblot showing Krüppel-like factor 4 (Klf4) protein abundance in macrophages (J774.A1) after exposure to NOX or HYX for 24 h or 48 h; Klf4 protein was related to β-actin, which served as a loading control; densitometric data are displayed under the immunoblot; n=4–5 per group. e) Klf4 was suppressed in macrophages (J744.A1). Cells were transfected with small interfering RNA (siRNA) against Klf4 (si-klf4) or scrambled siRNA (scr) using the endo-porter technique, followed by a recovery time of 24 h in serum-rich medium. Subsequently, cells were starved with serum-reduced medium. Gene expression of macrophage markers was assessed after exposure to HYX for 48 h:白细胞介素6金属蛋白酶12 (Mmp12),toll样受体4 (Tlr47)和CC-chemokine配体(Ccl7);β-actin担任管家基因;均值±扫描电镜;n =每组4 - 5。f)主要小鼠巨噬细胞分离、培养(腹膜)24 h serum-rich中。随后,细胞暴露于氮氧化物或HYX serum-reduced 48 h的媒介。M1-like基因表达(Mmp12,Tlr4)和M2-like(精氨酸酶1 (__arg1),resistin-like分子α1 (Fizz1使用定量实时PCR)标记进行评估;β-actin担任管家基因。g)和(b)类似,主要小鼠巨噬细胞暴露在氮氧化物或HYX 48 h;随后MLE12受到HYX和CM处理氮氧化合物,CMHYXVeh(未经处理的介质,Co)的主要或小鼠巨噬细胞;控制MLE12暴露于氮氧化物。24小时后,扩散MLE12使用MTT分析评估;n =夫人每组。h)展示肺片14-day-old小鼠治疗与Veh或il - 6 / sIL-6R和暴露于氮氧化物或HYX 48 h;n = 3 - 4每组。代表co-immunofluorescence染色末端转移酶的dUTP尼克结束标签(TUNEL)作为细胞凋亡的指标(绿色)和表面活性剂蛋白C (SFTPC) (ATII、红色)×100放大,以及定量总结数据的TUNEL的百分比+和SFTPC+:TUNEL+细胞相对于所有ATII;均值±扫描电镜。配对t检验:*:p < 0.05, * *: p < 0.01, * * *: p < 0.001;重复的措施。单向方差分析后跟Bonferroni测试后:#:p < 0.05,# # # #:p < 0.0001。DAPI: 4′, 6-diamidino-2-phenylindole。
人类将我们的数据,我们使用hAECs (补充图S4人类monocyte-derived)和巨噬细胞。治疗hAECs与il - 6 / sIL-6R显著降低扩散normoxia和氧过多24 h (图7)。我们下一个暴露hAECs标准单元中(Co), normoxia-conditioned人类M1-like巨噬细胞中(hCM)氮氧化合物)或hyperoxia-conditioned人类M1-like巨噬细胞(hCM媒介HYX)。类似于小鼠的研究中,我们发现hCMHYX,但不是hCM氮氧化合物进一步降低扩散hAEC (图7 b)。有趣的是,厘米HYXM0-like巨噬细胞没有添加剂的影响相比hyperoxia-reduced hAECs扩散到CM氮氧化合物(补充图S5)。最后,暴露M1-like分化人类monocyte-derived巨噬细胞氧过多48 h显著增加人类的基因表达白细胞介素6(h白细胞介素6),TNFa,TLR4和IL1b(图7 c)。
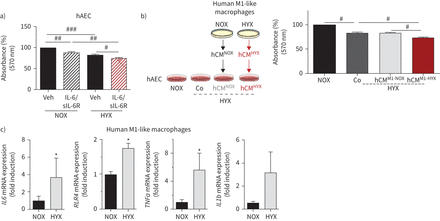
白介素6 (il - 6) /可溶性白介素受体(sIL-6R)和人类M1-like巨噬细胞影响人类肺泡上皮细胞(hAEC)体内平衡。a)扩散hAECs评估使用MTT试验;hAECs在serum-rich培养基培养24小时。之后的细胞治疗与车辆(阿明费)或il - 6 / sIL-6R和暴露于氧过多(HYX;85%啊2)或normoxia(氮氧化物;21%啊2为24小时);n = 6 /组。b)方案,描述了实验设计:人类M1-like巨噬细胞暴露在氮氧化物或HYX 48 h和条件培养液(CM)收集;随后,hAECs治疗下HYX人类NOX-exposed巨噬细胞(hCM的CM氮氧化合物),hCMHYX24小时或车辆(Co);控制hAECs暴露于氮氧化物。最后,扩散hAECs使用MTT分析评估;n = 4 /组。c)人类单核细胞从血液和孤立对待粒细胞巨噬细胞集落刺激因子(100 ng·毫升−1)serum-rich介质为7天。随后,细胞被维护在serum-reduced介质和暴露于氮氧化物或HYX 48 h,紧随其后的是评估巨噬细胞表达的基因编码的标记(白介素6 (白细胞介素6),toll样受体4 (TLR4)、肿瘤坏死因子α(TNFa)和interleukin1b (IL1b使用定量实时PCR));β-actin担任管家基因。均值±扫描电镜;n =每组4 - 5。重复测量单向方差分析与Bonferroni测试后:#:p < 0.05;# #:p < 0.01;# # #:p < 0.001;Wilcoxon配对t检验:*:p < 0.05。
炎症在早产儿血清生物标志物分析氧气和桶
等离子体的细胞因子多元分析早产儿决定macrophage-related细胞因子(调节激活,可能正常t细胞表达和分泌(咆哮),monocyte-chemotactic蛋白3 (MCP3) monokine-induced由γ干扰素(MIG))和il - 6 (图8)作为一个潜在的生物标记物,确定婴儿接受氧气治疗的风险发展桶。通过混合线性模型,婴儿发展到桶演示了一个纵向模式血浆细胞因子水平的变化,不同于在婴儿,没有出现桶(图8 b- e)。此外,我们进行了非参数拟合曲线,然后通过统计分析来确定桶之间的区别与non-BPD在32周和36周(图8 f)。我们发现在32周婴儿发展中桶interleukin-1受体拮抗剂(IL1RA)浓度的增加(p = 0.026),和一个趋势增加细胞间粘附分子(ICAM) (CD54;p = 0.075)。这些细胞因子是两极分化或由巨噬细胞分泌的。在36周,我们只发现一个温和但不显著海拔干扰素γ(p = 0.16)和巨噬细胞炎性蛋白α(CCL3;p = 0.15),可能由于小样本大小。这将需要验证通过前瞻性纵向研究。
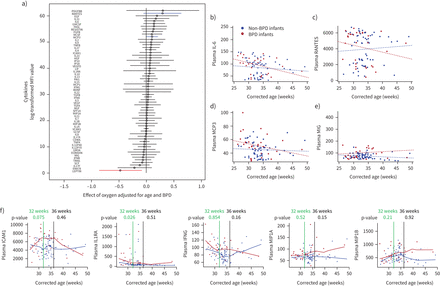
临床数据显示细胞因子浓度的动态等离子的婴儿有无支气管肺的发育不良(桶)。)森林图显示氧气在早产儿cystokine浓度的影响(n = 55)。数据是根据年龄和桶进行调整。使用多路检测血浆细胞因子测定;蓝色和红色线条突出最调节和细胞因子表达下调。小额信贷机构:平均荧光强度。抵扣)混合线性模型对纵向模式在婴儿血浆细胞因子水平的变化,发展到桶(红点)与non-BPD(蓝点)。血浆白介素6 (il - 6) (b);和巨噬细胞相关细胞因子(汉英):调节激活,可能正常t细胞表达和分泌(咆哮)(CCL5: CC-chemokine配体5)(c);monocyte-chemotactic蛋白3 (MCP3) (CCL7 CC-chemokine配体7日)(d);和monokine-inducedγ干扰素(MIG)(趋化因子(C-X-C主题)配体9)(e) f)非参数拟合曲线细胞间粘附分子1 (ICAM;CD54)、白介素1受体拮抗剂(IL1RA)、干扰素γ(IFNG)、巨噬细胞炎性蛋白α(MIP1A;CCL3;p = 0.15)和MIP1B(亚兰)。统计差异桶和non-BPD婴儿在其32周(绿线)和36周(黑线)。各自的假定值表示。
免疫(CD45+和CD68+)细胞激活的涌入il - 6 / STAT3信号和减少在肺上皮细胞与桶的婴儿
最后,我们分析了6个产后人类肺部桶和6个年龄控制肺(表1)。CD45 immunofluorescent染色显示在桶的肺部免疫细胞的显著增加。后续具体染色CD45和CD68,巨噬细胞的标记,显示明显高于肺的免疫细胞和巨噬细胞数量与桶相比non-BPD (图9,b)。原位杂交h白细胞介素6显示更多的h白细胞介素6mRNA-expressing细胞在肺部桶相比,年龄控制肺(图9 c)。的结合原位杂交h白细胞介素6和CD68 immunofluorescent染色显示的表达明显高于h白细胞介素6在肺巨噬细胞比non-BPD桶(图9 d)。il - 6的Immunofluorescent染色STAT3的下游,CHD1(上皮细胞标记)显示一个激活的STAT3和减少背景+细胞只有在性别和年龄对(男性),而婴儿年龄匹配,但不是性(男性与女)变化不显著,表明可能的性别差异(图9 e- h)。最后,我们染色ATII SFTPC作为标记,发现了一个老年协会的ATII桶。婴儿的年龄与减少ATII桶显著相关(R2= 0.5794;p = 0.0062),而non-BPD显示ATII随着年龄增加的趋势(图9我)。总之,这些数据表明,桶的发病机制与macrophage-derived h炎症有密切关系白细胞介素6,STAT3激活,最终失去上皮细胞和ATII。
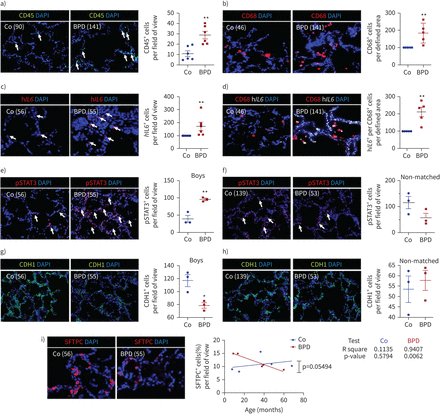
炎症和白介素6 (il - 6) /信号传感器和转录激活3 (STAT3)信号在肺和支气管肺的发育不良的婴儿(桶)和non-BPD。)代表immunofluorescent为免疫细胞染色用CD45标记(绿色)在肺与桶和控制(有限公司);婴儿的肺身份证号码在括号中表示。免疫细胞(CD45+细胞)数在4 - 12的视野/肺。摘要免疫细胞(CD45的量化数据+细胞)的视野对所有婴儿每组(n = 6)。DAPI: 4′, 6-diamidino-2-phenylindole。b)代表co-immunofluorescent本地化CD68(巨噬细胞的标志,红色)与桶和肺;白色箭头指示CD68+细胞。CD68的分析+细胞每6 - 12字段旁边的视图显示图像;n = 6 /组。c)代表人类的本地化白细胞介素6(h白细胞介素6,在肺和桶有限公司红色);白色箭头指示h白细胞介素6+原位杂交。h的分析白细胞介素6+细胞每6 - 12字段旁边的视图显示图像;n = 6 /组。d)代表co-immunofluorescent本地化CD68(巨噬细胞的标志,红色)和h白细胞介素6(白色;原位杂交)与桶和肺;白色箭头指示CD68+和h白细胞介素6+。h的分析白细胞介素6每CD68 mRNA表达+细胞每6 - 12字段旁边的视图显示图像;n = 6 /组。e、f)代表immunofluorescent染色显示在肺和桶有限公司pSTAT3阳性染色;白色箭头表示pSTAT3+细胞。分析pSTAT3+细胞4 - 12字段的视图进行性别和年龄桶和控制肺(男孩;每组n = 3) (e)以及年龄相仿,但不是sex-matched桶和控制肺部每组(n = 3) (f)。旁边的图形显示各自的图像。g h)代表immunofluorescent染色显示背景(绿色)阳性染色在桶和控制肺部。背景的分析+细胞4 - 12字段的视图进行性别和年龄桶和肺(男孩;每组(n = 3) (g)以及年龄相仿,但不是sex-matched桶和肺每组(n = 3) (h)。旁边的图形显示各自的图像。我)代表immunofluorescent表面活性剂蛋白C (SFTPC;标记的肺泡上皮II型细胞(ATII),红色)在桶和肺。线性回归的分析SFTPC的数量+和年龄所示图(桶,红色;有限公司、蓝);r2和假定值是表示在图;n = 5 /组。均值±扫描电镜。Mann-Whitney测试或学习任务:*:p < 0.05;* *:p < 0.01。
讨论
本研究提出了一个新颖的机制氧过多负面影响ATII体内平衡和弹性纤维的形成通过巨噬细胞活化,导致增强il - 6轴的激活白介素trans-signalling通过sIL-6R,最终形成抑制肺泡和肺功能损害。的数据提供了强有力的证据表明抑制il - 6 trans-signalling提供了一个创新的药理增长目标,使肺早产儿的风险桶。这部小说炎症在桶的发病机制是基于范例在活的有机体内研究利用基因改造小鼠与删除白细胞介素6,药物治疗针对全球和il - 6 trans-signalling,最后人类研究使用血清和肺的婴儿有或没有桶hAEC和monocyte-derived巨噬细胞细胞培养研究。
炎症是桶的中央机制(13]。最近的研究强调,居民由氧过多肺泡巨噬细胞被激活,导致它们有助于异常结构发展的不成熟的小鼠肺15]。Kalymbetovaet al。(15]表明,氧过多造成的损失居民肺泡巨噬细胞在新生鼠肺,而小说CD45人口+CD11c+SiglecF+CD11b+CD68+MHCII+细胞出现。作者建议住宅肺泡巨噬细胞transdifferentiate到这个新的人口,导致逮捕alveolarisation [15]。在目前的研究中,第一次,我们应用一个全面的生物信息学方法使用在网上反褶积的细胞比例从转录组数据在模型实验桶。我们确定一个免疫细胞比例降低t细胞和b细胞的数量增加肺的老鼠暴露于长期氧过多。这类似于最近的一份报告显示CD4阳性的重要消耗+以及CD8+t细胞(24后,天真的b细胞的比例增加氧过多(25]。最引人注目的,我们的数据确定减少M2-like巨噬细胞,并upregulation M1-like肺泡巨噬细胞(巨噬细胞可能出现的居民15]。进一步测试这个概念和定义不同的巨噬细胞表型单细胞RNA序列应该执行。
进一步的分析验证,趋化因子调节巨噬细胞功能是高度调节氧过多,cytokine-cytokine受体相互作用和IL-17信号是受影响最严重的途径。有趣的是,il - 6指导免疫反应对Th17表型,和增加IL-17氧过多后肺泡巨噬细胞和肺上皮细胞(26,27]。il - 6在新生儿肺损伤的作用,然而,一直备受争议的讨论。虽然超表达il - 6加重肺损伤在新生儿出生后小鼠暴露在氧过多,il - 6对oxidant-mediated超表达提供了保护效应损伤小鼠暴露于氧过多时从三到六天的生活(28,29日]。相比之下,目前的研究表明白细胞介素6零老鼠表现出轻微减少肺泡已经normoxia下可能与减少myofibroblasts和可能受损的次要的分隔作用。虽然这概念是投机,需要进一步的调查,我们目前的研究结果与之前的研究突出il - 6的严格监管的重要性在离散的窗户肺发展。
各种实验模型被用来研究桶的发病机制,包括暴露在氧气浓度增加,机械通风或感染(10,30.,31日]。解释数据时不同的标准,包括性别、年龄、应变、有害刺激和持续时间的接触必须考虑(32- - - - - -34]。报告表明,损伤和肺表型取决于有害刺激和曝光时间。例如,连续暴露于40%2从P1到P4或好不利影响肺泡表面形成和气体交换,但不间隔的壁厚作为指标矩阵改造。相反,只有严重的桶模型与接触新生幼鼠身上85%的O2戒律都显著影响alveolarisation和肺泡间隔壁厚[32,35,36]。在目前的研究中,我们使用85% O2长期诱导炎症严重的慢性肺损伤明显。然而,未来的研究应该包括氧浓度的影响和曝光时间结合机械通风和/或感染。
il - 6是一个多功能的促炎细胞因子,调节细胞生存、分化的免疫细胞,如巨噬细胞、中性粒细胞以及t细胞,诱导矩阵改造,有利于纤维化(37,38)如慢性肺疾病(39]。临床研究显示il - 6的浓度增加,sIL-6R和sgp130气管吸入的婴儿进化桶(40- - - - - -42]。ATII-macrophage交互提出了之前的研究显示,肺泡上皮IL-6-activated STAT3信号介导炎症,可能通过巨噬细胞(43]。我们提供证据在活的有机体内和在体外氧过多会影响macrophage-hAEC交互通过增加不同的巨噬细胞调节细胞因子,包括白细胞介素6,诱导减少hAECs的生存,促进M1-like分化的巨噬细胞44,45]。因为先前的研究表明,减少Klf4赞成M1-like分化,可能我们现在的结果可能与损失Klf4后氧过多(23]。总的来说,我们的数据突出氧过多的程度也许肺泡上皮的巨噬细胞,但也在策划这种细胞il - 6的关键作用相声。最近的报告表明,针对il - 6信号阻止emphysema-like疾病(46),可能是一个治疗肺动脉高血压的新目标(47]。在这里,我们提供的证据表明,il - 6和经典药物抑制il - 6, trans-signalling使肺泡生长和肺功能通过保护ATII生存和保持弹性纤维分布在新生鼠肺氧过多。最近的研究表明,抑制蛋白水解活性的机械通风使用丝氨酸弹性蛋白酶抑制剂抑弹性酶蛋白使新生鼠肺增长和减少炎症(21,30.]。蛋白酶的关键作用,在新生儿肺anti-protease平衡增长支持的进一步发现婴儿进化桶增加血清弹性蛋白酶浓度(30.]。有趣的是,蛋白酶(如。ADAM10或ADAM17)描述了作为sheddase白介素受体和监管者的il - 6信号(48- - - - - -50]。因此,阻断蛋白水解活性可以通过阻止il - 6信号抗炎效果。
炎症反应和细胞因子的调节异常是桶的突出特征。桶的免疫细胞数量增加与减少分隔作用和更少的肺泡12,13,15]。在目前的研究中,我们决定增加血浆细胞因子的浓度调节巨噬细胞招聘和功能,如咆哮或MCP3,突出系统性炎症反应桶。同样地,我们发现一个il - 1受体拮抗剂和ICAM1的临床upregulation细胞因子调节巨噬细胞分化[51,52]。有趣的是,血清ICAM水平最近报道作为桶的潜在生物标志物,并与它的严重性(53]。血浆il - 6浓度与il - 6的早期产后海拔动态监管趋势的婴儿发展桶non-BPD相比。由于等离子体不一定反映lung-intrinsic il - 6,气管吸入物可能更适合在先前的研究中所示增加il - 6与桶(婴儿42]。然而,测量的时间点的婴儿可能不匹配我们的小鼠模型。血浆细胞因子模式与桶婴儿与免疫细胞的显著增加,特别是巨噬细胞(CD68+),macrophage-derived增加白细胞介素6在肺的婴儿桶,独立的性别与年龄。我们联系这个免疫反应激活STAT3信号和失去上皮细胞的男性婴儿。有趣的是,在与这些影响并不明显,但不是sex-matched,桶和non-BPD婴儿,暗示可能性别il - 6 / STAT3的反应,这可能有助于sex-dependent桶的差异(54]。最后,我们显示出年龄和减少之间的相关性肺ATII的桶,但不是在non-BPD。总的来说我们的研究,与之前的研究一致,表明macrophage-regulating细胞因子il - 6,可能会成为一个有用的肺损伤的生物标志物早产儿,并帮助定义桶的风险。
中所描绘的一样我们的工作模型(图10),炎症和损伤的再生上皮利基ATII损失损害肺泡形成并导致气性肺结构之外的初级阶段。目前的研究表明氧过多会破坏macrophage-ATII交互通过触发il - 6 / STAT3信号在小鼠和人类肺,还确定了il - 6作为一个潜在的早发性生物标志物和sgp130Fc婴儿作为新的治疗策略的风险桶。il - 6的具体封锁trans-signalling主要抑制il - 6的炎性活动而保护和再生功能不受影响。这种创新的治疗策略已经在二期临床试验患者的自身免疫性疾病(55)和桶的治疗提供了一条新的道路。
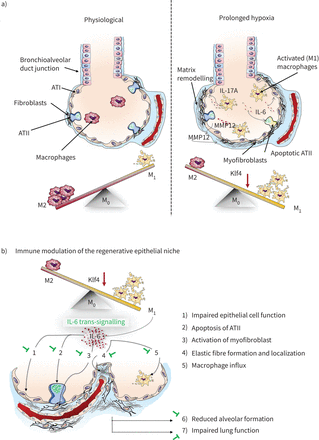
提出工作模型说明如何氧过多诱发巨噬细胞分化。一)M1-like-driven炎性表型导致细胞因子的分泌,如白介素6 (il - 6)和IL-17A,巨噬细胞弹性蛋白酶,也称金属蛋白酶12 (MMP12)。这个炎症肺泡微环境减少肺泡上皮的生存II型细胞(ATII),而成纤维细胞被激活和矩阵改造提升。ATI:肺泡上皮型细胞;Klf4: Kruppel-like因素4。b)两极分化的巨噬细胞氧过多会导致释放il - 6后,负面影响再生上皮利基。然而,il - 6缺乏保护从这些变化1)促进表面活性剂蛋白质的表达和2)生存ATII, 3)减少myofibroblasts, 4)保护从摄动弹性纤维组装和本地化,和5)衰减巨噬细胞大量涌入。这些有益的影响il - 6在肺泡间缺乏使6)肺泡形成和7)从降低肺功能保护,提供一个新的治疗目标治疗桶。
补充材料
可共享的PDF
确认
我们非常感激和感谢玛琳因和理查德乏味(儿科部门,心血管病研究所,维拉·默尔顿墙肺血管疾病中心,斯坦福大学医学院,斯坦福大学)的有用的建议。我们感谢科隆卓越的成像设备集群在Aging-Associated疾病的细胞应激反应(CECAD)为他们的技术支持。我们还要感谢Sebahattin Cirak(儿科部门、分子医学中心科隆和罕见疾病中心科隆科隆大学、科隆大学医院,科隆,德国)和Ugur Akpulat(儿科部门、分子医学中心科隆科隆大学、科隆大学医院,科隆,德国;和大学该国中北部毗邻黑海的卡斯塔莫努,土耳其)在细胞培养研究的技术支持。Stefan Rose-John IL-6mAb和sgp130Fc请提供(生物化学研究所Christian-Albrechts-University基尔,德国)。
脚注
可以从本文的补充材料www.qdcxjkg.com
作者的贡献:d . Hirani Alvira, s . Danopoulos c·米拉·d·艾尔·阿拉姆,诉Barbarino, c . Pallasch美国玫瑰约翰,m . Odenthal 9 Pryhuber, r .岛上和硕士Alejandre城堡的构思和设计研究;d . Hirani Alvira, s . Danopoulos m . Donato c·米拉k全垒打,c . Vohlen j .莫尔诉Barbarino Mansouri和硕士Alejandre城堡进行实验;d . Hirani Alvira, s . Danopoulos m . Donato c·米拉l, c . Vohlen d·阿尔·阿拉姆和硕士Alejandre城堡分析数据;d . Hirani Alvira, s . Danopoulos m . Donato c·米拉l .田j . Selle s . v . Koningsbruggen-Rietschel j .莫尔·Khatri d·艾尔·阿拉姆Dotsch和硕士Alejandre城堡解释实验结果;d . Hirani s Danopoulos m . Donato还和硕士Alejandre城堡准备数据;d . Hirani和硕士Alejandre城堡起草的手稿;d . Hirani Alvira, s . Danopoulos c·米拉·d·艾尔·阿拉姆,j . Selle w·格和硕士Alejandre城堡编辑和修改手稿;d . Hirani Alvira, s . Danopoulos m . Donato c·米拉l .田j .莫尔k全垒打,c . Vohlen j . Selle s . v . Koningsbruggen-Rietschel诉Barbarino, c . Pallasch美国玫瑰约翰,m . Odenthal 9 Pryhuber, r .岛上s Mansouri w·西格,p . Khatri d·艾尔·阿拉姆Dotsch和硕士Alejandre城堡批准最终版本的手稿。
利益冲突:d Hirani没有披露。
利益冲突:蔡玫Alvira没有披露。
利益冲突:美国Danopoulos没有披露。
利益冲突:c·米拉报告赠款Proteostasis (aapl . o:行情)和Eloxx制药、赠款和个人费用从顶点药品顾问委员会的工作,在提交工作。
利益冲突:m . Donato没有披露。
利益冲突:l .田没有披露。
利益冲突:j .莫尔没有披露。
利益冲突:全垒打没有披露。
利益冲突:c . Vohlen没有披露。
利益冲突:j .还没有披露。
利益冲突:美国诉Koningsbruggen-Rietschel没有披露。
利益冲突:诉Barbarino没有披露。
利益冲突:c . Pallasch没有披露。
利益冲突:美国玫瑰约翰没有披露。
利益冲突:m . Odenthal没有披露。
利益冲突:9 Pryhuber报告从NHLBI赠款,在进行这项研究的。
利益冲突:美国Mansouri没有披露。
利益冲突:r .岛上没有披露。
利益冲突:w·西格报告个人费用从Actelion股价,Abivax,拜耳公司,Vectura, Medspray,联合疗法,Liquidia地区,在提交工作。
利益冲突:p . Khatri没有披露。
利益冲突:d·艾尔·阿拉姆没有披露。
利益冲突:j . Dotsch没有披露。
利益冲突:硕士Alejandre城堡没有披露。
支持声明:这项工作是由德意志Forschungsgemeinschaft 1636/2-1;硕士Alejandre城堡),河南和沃尔特·鲍尔Stiftung(硕士Alejandre城堡);科隆分子医学中心(硕士Alejandre城堡),和科隆大学医院;斯坦福大学的儿童健康研究所Tashia和约翰Morgridge教师学者奖(蔡玫Alvira);美国国立卫生研究院HL122918(蔡玫Alvira), NIH R01HL141856 (d·艾尔·阿拉姆),NIH po1hl108797 - 5974生物标志物核心(c .米拉);NHLBI LungMAP研究中心。U01HL122681 (s . Danopoulos d·艾尔·阿拉姆),Biorepository (1 u01hl122700, 9 Pryhuber)和数据协调中心(1 u01hl122638 9 Pryhuber)。资金信息,本文已沉积的Crossref资助者注册表。
- 收到了2020年6月12日。
- 接受2021年6月24日。
- 版权©2022年作者。
这个版本分布在创作共用署名非商业性许可证的条款4.0。商业生殖权利和权限接触权限在}{ersnet.org











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}