





文摘
尽管有证据表明传输驱动是一个广泛耐药结核病(xdr - TB)流行,我们理解之间的位置和人发生传播是有限的。我们试图确定是否存在个体间基因传播的证据没有流行病学联系。
我们进行了一项前瞻性研究广泛耐药结核的病人在夸祖鲁-纳塔尔省,南非,2011 - 2014年期间。我们收集社会人口和临床数据,确定了基于人际或医院流行病学联系联系。我们进行了全基因组测序(WGS)结核分枝杆菌隔离和确定两两单核苷酸多态性(SNP)差异。
404名参与者中,123例(30%)有叫人或医院联系,留下281(70%)流行病学链接。中位数SNP参与者与人与人之间的差异和医院联系10(四分位距(差)日到24日)和16(差10-23),分别。中间分离的参与者和他们最亲密的基因组SNP区别联系5(差3 - 9)和链接的一半参与者在7个SNP的至少五个参与者。
大多数epidemiologically-unlinked广泛耐药结核患者低成对SNP差异与至少一个其他参与者,符合传播。传播这些数据表明,可能由于偶然接触在社区设置个人之间彼此不认识。
文摘
广泛耐药结核的传播可能源于偶然接触个体之间彼此不认识http://ow.ly/4Wqt30lnHtp
介绍
耐药结核病(TB)在许多国家正在增加,并可能扭转最近在结核病控制(1,2]。治疗耐药结核病涉及复杂,有毒的和昂贵的治疗方案,与高死亡率和糟糕的结果(2]。鉴于这些挑战,疾病预防耐药以减少发病率和死亡率是至关重要的。尽管越来越多的证据表明,传播导致耐药结核病的传播在世界的许多地方3- - - - - -9),我们的理解和他们之间传输的发生仍然有限。这些我们在知识方面的排除能力设计有效的干预措施停止传输,防止耐药结核病新发病例。
从历史上看,接触调查被用来描述结核病疫情和传播链。分子基因分型的出现后,结核分枝杆菌strain-level数据补充和增强联系调查,这样情况下具有类似菌株被假定是传输网络的一部分(10]。然而,结核病传播的大量研究发现,流行病学和基因型的数据并不总是一致。例如,连接近距离接触可以确定只有9 - 30%的genotypically-linked个人通过一系列的设置(11- - - - - -14),这表明个体间传输可能源于偶然接触不知道。同样,家庭联系发病率高设置的研究已经发现,多达半数的次生结核病例不遗传型的与他们的假定指数情况下11,15]。这些发现可能反映了压倒性的疾病负担和大量的传输机会在结核病流行的地区。之间的这种不和谐密切接触者中流行病学和基因型的数据也表明,额外的工具,除了接触调查和传统的基因,可能需要确定大多数传播事件,特别是在高负担的设置。
全基因组测序(WGS)允许更精确描述结核菌株之间的遗传差异大于90%的通过检查结核分枝杆菌基因组,与传统pcr相比,不到1%。WGS标识顺序单核苷酸多态性(SNP)积累差异和促进了传动链的建设,而不是简单的通过基因分型确定集群相关的案件。此外,个人很少有SNP他们之间的区别结核分枝杆菌菌株可能代表一个传输连接(16,17]。
我们最近表明,69 - 92%的广泛耐药结核病(xdr - TB)在夸祖鲁-纳塔尔省,南非可以归因于人际传播耐药菌株,而不是收购的抵抗结核病治疗之前的设置(7]。我们发现通过密切接触或流行病学联系住院治疗上广泛耐药结核病例的30%,而流行病学来源不确定剩余的70%的病例。这些发现类似于从以前的以人群为基础的传播研究估计,推断的社区传播缺失的基因型或基因传播之间的联系密切接触者(11- - - - - -15]。然而,之前的研究没有仔细检查基因传输链接个人没有一个流行病学联系。在这项研究中,我们利用WGS识别潜在传播之间的联系不密切接触或重叠的住院病例。通过集成流行病学和基因组数据,我们将演示如何WGS可以提高我们的能力来识别广泛耐药结核传播事件发生的偶然接触个体之间彼此不认识。更好地了解社区传播会通知中断传播的努力目标,加快持续的努力减少全球结核病负担的。
方法
研究背景和人口
这个研究是在夸祖鲁-纳塔尔省进行,有人口1030万人,结核病的发病率最高(每000人口100 1076例)和艾滋病毒患病率(16.9%)在南非18,19]。广泛耐药结核患者culture-confirmed驻留在夸祖鲁-纳塔尔省招募了艾滋病的传播XDR结核(干什么)研究从2011年到2014年7]。TRAX研究的主要目的是确定患者的比例获得广泛耐药结核(即。二次治疗)不足与广泛耐药结核病的传播。
广泛耐药结核病例通过单一参考实验室进行药敏测试所有公共医疗设施在夸祖鲁-纳塔尔省。鉴于大量广泛耐药结核病例诊断,每年的便利样本诊断患者筛查招生。使用参考实验室数据库,年龄、性别和地区的参赛者和unenroled参与者之间的比较来确定研究样本的代表性。书面知情同意从所有参与者获得,或者从死者的近亲或重病的参与者。面试进行了收集参与者社会人口,结核病和艾滋病的历史,所有住院治疗上的位置和持续时间前5年。社交网络采访了在家密切接触者的信息,工作和其他地方参与者至少花2 h·周−1在前5年。详细的临床、社交网络和实验室一直在前面描述的方法(7]。
实验室方法
获得了广泛耐药结核的诊断孤立为所有参与者和recultured Lowenstein-Jensen偏。测序获得的材料进行人口文化板块的清洁工。基因组DNA提取和插入顺序6110年的限制性片段长度多态性(RFLP)基因分型结果进行根据标准方法(20.]。隔离了paired-end WGS并使用Nextera DNA测序图书馆准备工具包(Illumina公司、圣地亚哥、钙、美国)。生paired-end测序读Illumina-MiSeq平台上生成(美国Illumina公司,圣地亚哥,CA)和对齐H37Rv参考基因组(NC_000962.2)使用burrows - wheeler对准器[21]。隔离所读取覆盖>参考基因组的99%,最低的是深度报道任何隔离15 x。snp检测使用标准成对重测序技术(Samtools版本0.1.19)对质量的参考和过滤,读共识交替等位基因(> 75%)和接近indels从任何indel(少于50个碱基对)。单核苷酸多态性在或在50个碱基对的高变的Pro-Pro-Glu (PPE) / Pro-Glu (PE)基因家族,重复区域和移动元素也被排除在外22]。
社会网络分析
我们分析了社会网络采访数据识别参与者之间的流行病学联系。人与人之间的联系被定义为两个参与者命名或命名为一个共同的中介。我们确定了医院流行病学联系如果有重叠住院日期至少一个参与者在一个“脆弱的时期,”定义为至少1个月前收集的广泛耐药结核的诊断标本。一些参与者有多叫人和/或医院的链接。参与者没有叫人或医院链接被认为是”链接。“社会人口、临床和社会网络数据连接和分离的参与者相比,使用卡方或克鲁斯卡尔-沃利斯测试。
全基因组序列分析
WGS数据链接和链接的参与者相比,我们感兴趣的能力WGS基因分型结果提供进一步的歧视之外,我们专注于SNP参与者之间的差异与匹配的RFLP图案(定义为在一个乐队)。因此,对于相关的参与者,我们发现那些与他们匹配的RFLP图案流行病学联系然后成对SNP这些个体之间的差异决定的。如果一个参与者有多个流行病学与匹配的RFLP图案,我们选择的联系最少的成对SNP差异(即。“亲密”)为了专注于最有可能反映了传播的联系。
分离的参与者,我们发现他们最亲密的基因组研究队列内链接使用成对SNP的差异。使用这种方法,分离的参与者有机会被连接到所有其他参与者,这将增加他们的基因组与低概率的SNP仅靠机会的差异。为了考虑这种可能性,我们进行了敏感性分析来评估是否链接参与者多个基因连接在SNP阈值符合传播,这将支持传播的可能性与至少一个其他参与者没有一个流行病学与他们联系。为每个链接参与者我们确定其他参与者的数量在五,七,或10个snp。
道德的考虑
机构审查委员会批准的这项研究是埃默里大学的阿尔伯特·爱因斯坦医学院和夸祖鲁-纳塔尔大学和美国疾病控制和预防中心。
结果
研究人群
2011年5月到2014年8月,1027名患者被诊断出患有culture-confirmed广泛耐药结核在夸祖鲁-纳塔尔省(图1)。研究人员接洽和筛选521 culture-confirmed广泛耐药结核患者和404年(78%)合格,同意报名学习。参与者的参赛者从每个夸祖鲁-纳塔尔的11区和代表,按年龄,性别和地理,广泛耐药结核病例诊断在研究期间全省范围(分别为p = 0.52, 0.76和0.70)。的参与者中,234(58%)是女性和311年(77%)被感染艾滋病。34岁年龄中位数(四分位范围(差)28-43)(表1)。
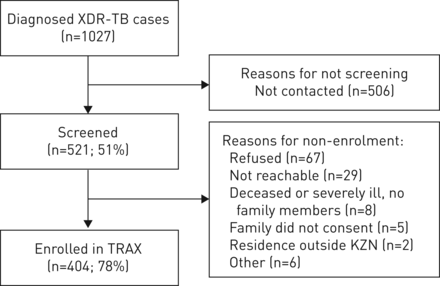
为参与者报名流程图。广泛耐药结核:广泛耐药结核病;TRAX:艾滋病XDR结核的传播研究;我们吃:夸祖鲁-纳塔尔。
社会互动和机动性
我们探索了参与者的社会互动和流动性来洞察他们的接触机会和结核病的传播。参与者叫共有2901名密切接触者从他们的家庭,工作场所和其他社区的地点,和一个叫每个参与者的平均7个联系人(差4到10)(表2)。在外工作报告了123名参与者(30%)和129名参与者(32%)报告支出> 2 h·周−1在一个社区聚集位置(如。教堂、酒吧和美发沙龙)。46个参与者(12%)报告使用公共交通超过1 h·天−1在报名之前。在5年之前,他们广泛耐药结核诊断,298人(74%)报告至少一个住院,86人(29%)报告两个或两个以上的住院治疗上。这些住院治疗上发生在53个不同医院,3个月的平均入学(差2 - 5)。
流行病学的链接
有59个参与者(15%)与一个叫人链接到至少一个其他参与者。链接的多数(84%)的家庭成员,虽然链接也包括同事(7%)和其他个人在社区(9%)。参与者住院脆弱时期广泛耐药结核诊断之前,72年(18%)重叠与另一项研究的参与者和医院联系。参与者重叠的中间三个其他参与者(差队)。总的来说,流行病学联系被确定为123人(30%),其中8例(2%)有人际和医院联系。
剩余的281名参与者(70%)流行病学”链接。“这些分离的参与者没有显著不同于有关参与者关于社会人口和临床特点,除了有点老,也不太可能报告咳嗽(表1)。链接数量的参与者也没有差别密切接触者他们命名,他们住在城市还是农村,或他们的住院治疗上的数量(表2)。
基因组的链接
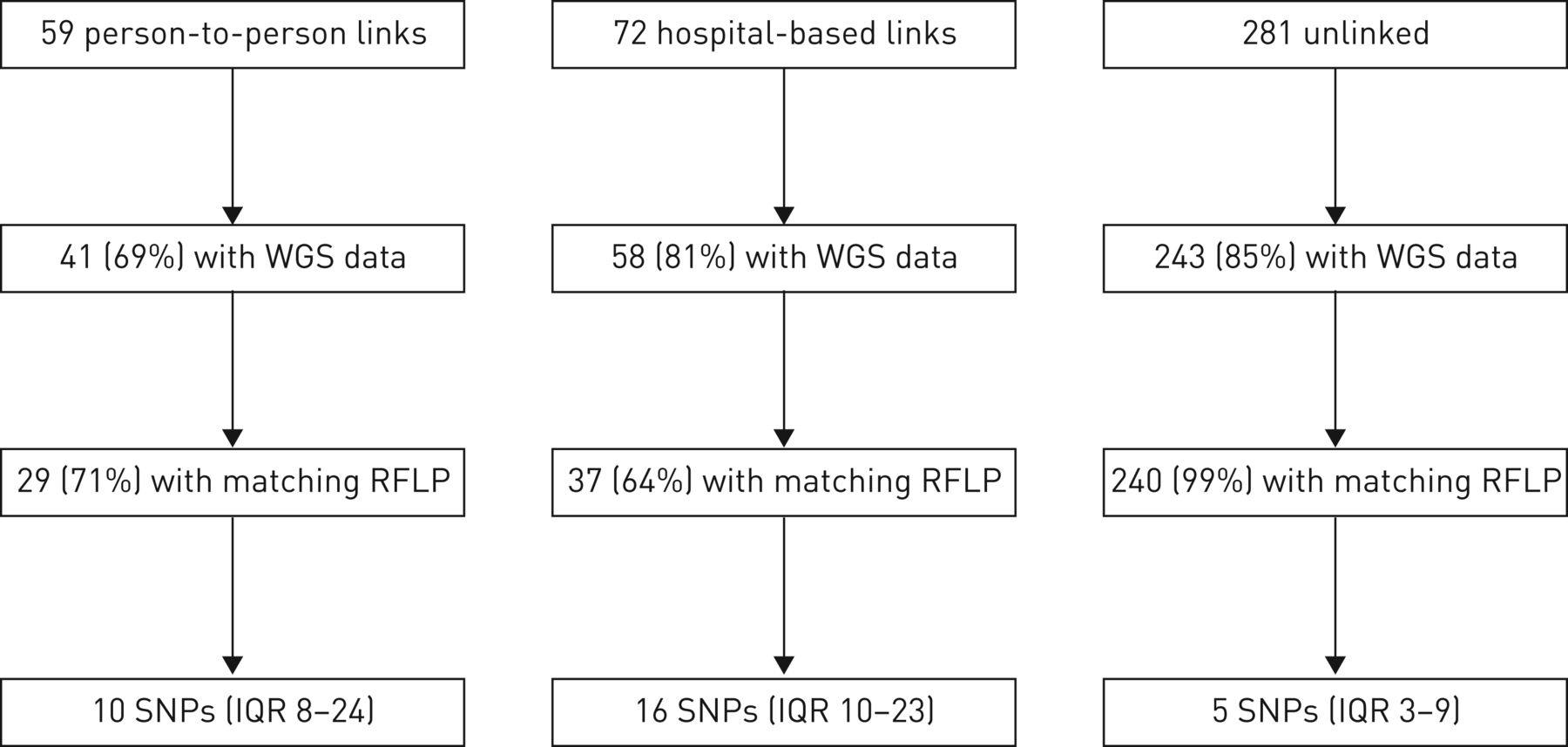
是6110年RFLP基因分型是完成结核分枝杆菌隔离的386名参与者(96%)和WGS 342年成功分离株(85%)(见补充表S1供单核苷酸多态性和参与者的特征补充表S2为RFLP集群)。其中包括41个59的参与者与人与人之间的链接(69%),58岁的72名参与者(81%)与医院联系281年和243年分离的参与者(86%)(图2)。
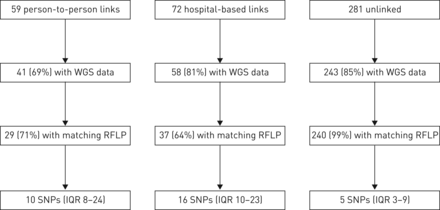
流程图的全基因组测序(WGS)数据的可用性和单核苷酸多态性(SNP)中位数差异的参与者,没有流行病学联系。RFLP:限制性片段长度多态性;差:四分位范围。
人与人之间联系的实验对象中,29日的41个(71%)有一个匹配的RFLP图案流行病学联系和中值成对SNP差异10 (IQR日到24日)(图2)。医院流行病学联系的实验对象中,37例(64%)有一个匹配的RFLP图案和中值成对SNP区别他们最亲密的医院联系16岁(差10-23)。epidemiologically-unlinked参与者,成对地影响他们最亲密的一对中值为5个snp(差3 - 9);因此,链接的一半参与者在5个snp的至少一个其他参与者。(见补充表S3SNP差异根据耐多药结核病的历史。)
单核苷酸多态性的分布差异提供了进一步支持分离的参与者之间的潜在传播事件图3)。SNP差异为链接参与者达到1 - 4单核苷酸多态性(47%),与一个额外的29%的参与者与5 - 9 SNP的差异。的结核分枝杆菌应变五分离的参与者是基因相同的另一个参与者(即。0 SNP差异)和18名参与者有一个链接结核分枝杆菌压力只有1 SNP不同于另一个参与者。相比之下,国民党不同参与者的人际和医院联系双峰分布,与一个高峰在5 - 9个SNP,另> 15单核苷酸多态性。这种双峰分布杜绝传输不同的SNP的识别阈值,作为已经被其他研究报告(11,23]。尽管如此,这个分布支持大多数链接参与者的可能性研究样本内的传输链路,因为78%的人在10个snp的至少一个其他参与者。
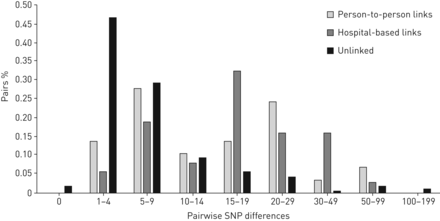
成对单核苷酸多态性(SNP)分布差异的参与者一个匹配的限制性片段长度多态性(RFLP)模式与基于名称的流行病学链接(n = 29),医院流行病学联系(n = 37),没有一个流行病学联系(即。分离的参与者,n = 240)。
进一步检查潜在的传播活动中分离的参与者,我们检查了基因连接的数量每个链接的参与者都有低于SNP阈值之前提出的暗示传输(23- - - - - -26]。共有192个链接参与者(79%)与至少一个其他参与者10或更少的单核苷酸多态性和中间的连接数量与29参与者在这个阈值(差1 - 80)(表3)。阈值≤7个snp, 173链接参与者(71%)与至少一个其他参与者和中间的连接数量在这个阈值是与其他五个参与者(差0-24)。最后,在一个阈值≤5个snp, 143年分离的参与者(59%)基因与至少一个其他参与者和中值的连接数与另一个参与者(IQR 0 - 9)。因此,近60%的参与者没有流行病学联系有基因联系符合传播的相对严格的阈值5个snp,许多参与者有多个链接在这个阈值。
讨论
南非面临耐多药结核病(mdr - TB)和广泛耐药结核病的流行由耐药菌株的传播。传播耐药结核病流行的主导作用已被证明在许多设置和模拟数据显示,传输将燃料在未来几十年全球耐多药结核病和广泛耐药结核病的增加(3- - - - - -6,27]。不幸的是,我们能够设计有效的公共卫生干预措施停止传播阻碍了我们有限的理解。在这项研究中,我们综合流行病学和基因组广泛耐药结核患者的最大队列中的数据日期和识别基因组链接传播暗示对于大多数epidemiologically-unlinked参与者。
研究参与者有许多广泛耐药结核传播的机会,与多个社会接触,经常参加社区聚集地点,住院治疗上和使用公共交通工具。在关注epidemiologically-unlinked参与者,我们惊奇地发现,他们中的大多数已经SNP差异相似甚至低于epidemiologically-linked参与者之间。事实上,我们发现,许多分离的参与者被连接到数个研究参与者只有几单核苷酸多态性。这些发现表明,这些参与者传输链接不会被传统接触调查或医院感染控制程序,但相反,源于休闲,以社区为基础的联系。随意的联系人之间的传递这些链接提供经验证据,支持未来的研究需要描述和量化结核传播在社会的位置。
我们的发现扩展一些先前的研究提出对社区传播的角色后,发现占少数的高负担的传播密切接触者设置(11,15,28- - - - - -30.]。在马拉维,使用基于WGS显示只有9.4%的传播发生在密切接触者(11]。研究从中国和来自南非的一项小型研究还发现,很大一部分genomically-linked病例没有流行病学联系(23,30.,31日]。然而,我们的研究提供了基因组epidemiologically-unlinked个人之间传播的证据,与多个基因的链接对于大多数患者加强司机的偶然接触传播的可能性。
近三分之一的epidemiologically-linked参与我们的研究有不同的RFLP模式,这与之前的研究一致,39 - 62%的家庭接触者有不同的菌株(11,12,15]。此外,SNP差异epidemiologically-linked参与者表现出双峰模式,一半的双有SNP的差异低于10 - 12个SNP另一半有SNP差异和15—50岁SNP范围,即使他们共享一个RFLP图案。这些发现,类似于在其他设置(11,15),建议,只有一小部分的二次例归因于与一个已知的指示病例密切接触。剩余的二次感染广泛耐药结核病例可能从其他的人密切接触。因此,在一个高负担设置如南非,一个流行病学链接的存在可能更代表共同危险因素暴露比真正的传输链路。
WGS被用于各种设置,以确定一个SNP阈值指示性传播(16,17),较低的SNP与流行病学联系个体间的差异被认为是“黄金标准”传播的几项研究[11,23]。然而,在我们的研究中,许多epidemiologically-linked参与者SNP差异高于先前提出的阈值(如。他一直SNPs) (17),无论是epidemiologically-linked还是epidemiologically-unlinked参与者SNP分布具有明显低于传输可以被视为可能的过渡点。定义的挑战SNP阈值进一步突出了最近的一项研究在伦敦的结核病疫情,14年间,将近60%的菌株不同零个或一个SNP和单核苷酸多态性之间的344名患者的最大数量是五(26]。进一步的研究需要阐明如何结核分枝杆菌根据病原体变异率不同,主机和流行病学因素。
WGS的解释数据有局限性,其中很多并不是特定于这对基因流行病学研究但代表广泛的挑战。例如,尽管特征变异率一直在实验室设置,它仍然是很难估计变异率从临床和流行病学数据的变量潜伏期结核和固有的不确定性,当一个人可能被感染,尤其是在高负担。同样,越来越多的文献表明在宿主的可变性结核分枝杆菌压力,在一个时间点,随着时间的推移17,32,33]。这个变化的临床意义,它是如何影响宿主因素,如艾滋病毒合并感染和传播,任何潜在的影响目前尚不清楚。克隆的影响结核分枝杆菌菌株,如LAM4 /我们应变在夸祖鲁-纳塔尔省[34),在传播动力学也不清楚。然而,SNP差异在这个群的多样性表明,WGS拥有足够的特异性分化潜在传播事件,即使在一个流行的存在压力。
不完整的捕捉限制我们的能力来识别所有传播事件,虽然我们的研究不是为了捕获所有在研究期间广泛耐药结核病例,可能更大的采样的数量将会增加流行病学联系和影响我们的估计SNP参与者之间的区别。然而,研究与全球更完整的抽样(如。美国、西班牙、荷兰和马拉维)还发现了一个高比例的情况下没有流行病学联系(11,12,14,35]。此外,我们发现大部分的链接参与者多个基因组链接,即使在严格的SNP阈值。因此,我们的研究结果可能代表的比例最低的估计归因于偶然接触传播。
的继续传播耐药结核病全球结核控制构成了严重威胁。我们停止这种日益流行的能力将铰链的设计有针对性的干预措施,阻断传播。我们发现许多偶然接触机会和个体之间的传播和广泛耐药结核。大多数的这些人还流行病学链接的人基因传输与其他参与者的证据,强调偶然接触的潜在重大贡献在夸祖鲁-纳塔尔省广泛耐药结核病的流行。虽然接触调查预防院内传播和感染控制计划已经证明受益的基本支柱结核病控制活动,进一步调查通过偶然接触传播的以社区为基础的设置必须决定如何以及在哪里进行干预和扩大我们现有的结核病控制的方法。
补充材料
确认
我们感激在夸祖鲁-纳塔尔大学的研究团队的不懈努力在数据收集、记录抽象,参与招聘和面试。我们感谢的参与者和他们的家人同意参与这项研究。
脚注
可以从本文的补充材料www.qdcxjkg.com
利益冲突:没有宣布。
支持声明:本研究主要是由格兰特R01AI089349(π天然橡胶甘地)从美国国家过敏症和传染病研究所(NIAID) /国家卫生研究院(NIH)。也支持部分由NIH / NIAID赠款R01AI087465(π天然橡胶甘地)K23AI083088(πJ.C.M. Brust), K23AI134182(π南卡罗来纳州老的)和K24AI114444(π天然橡胶甘地),由艾莫利CFAR P30AI050409(πj . Curran),被爱因斯坦CFAR P30AI051519(πh . Goldstein),由爱因斯坦/蒙特两个UL1 TR001073(πh . Shamoon)和由NIH /国家心脏,肺和血液研究所(NHLBI)授予T32 HL116271(πd . Guidot)。研究结果和结论在这个手稿的作者和不一定代表官方立场的疾病控制和预防中心或美国健康和人类服务。资金信息,本文已沉积的Crossref资助者注册表。
- 收到了2018年2月6日。
- 接受2018年8月8日。
- 版权©2018人队











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}