





摘要
虽然IFN-gamma表达增强仍然是结节病的主要特征,但其起源可能涉及Th1以外的因素http://ow.ly/qtyB30i48bU
免疫学上,结节病是通过本地归因于疾病部位的Th1-显性环境的典型定义,其特征在于干扰素(IFN)γ和Th1促进白细胞介素(IL)12和IL18 [1]。因此,在结节病变中也观察到增强Th1的下游效应,例如Th1相关趋化因子(MIG / CXCL9,IP10 / CXCL10,ITAC / CXCL11,MCP1 / CCL2,MIP1α/ CCL3,MIP1β/ CCL4,Rantes /CCL5)及其各自的受体(CXCR3,CCR5)。相反,增强TH2的存在从未在结节病肺中牢固地记录。
最近的凌志是b鲁斯等.[2有助于对这些经典的观察进行不断的改进。通过评估th1相关和th17相关趋化因子受体的表面表达,他们证明具有Th17.1表面表型的t细胞主要存在于结节病肺(支气管肺泡灌洗)和邻近纵隔淋巴结(MLN)。使用这种方法,它们也出现在表面(不是双关语),以确定结节病肺中Th1表型的减少和Th1与临床状态(缓解)缺乏相关性与慢性疾病)。事实上,他们的研究将Th17.1与慢性活动性结节病联系在一起,作者提出检查Th17.1/Th1比值可以作为疾病病程的预后指标。
然而,作者也引用了他们的数据与Kais等.[3.[据报道,据报道,Th17.1与经历Löfgren综合征(急性结节病)的斯堪的纳维亚患者疾病缓解有关。这两个手稿之间的差异不太可能由患者群体引起临床表型和遗传学的患者,而是用于定义辅助T细胞亚群的方法。
斯堪的纳维亚研究检测了与Th1和Th17功能相关的转录因子的表达,分别是Tbet和RORγt [3.]。从这种观点来看,测量TBET +RORγT+的细胞内表达+在细胞水平下定义了更高的TH17 / TH1比。这些细胞表达了较高的IL17和较低的IFNγ,这种功能表型与急性结节病患者的患者有利的疾病结果相关。这种观察似乎与接受的教条相一致,与IFNγ的表达相关的结节病相关的临床终点相关[4.]。
其中介于差异:通过功能定义辅助T细胞子集(kais等.[3.])与通过表面表型(B鲁斯等.[2])。虽然Th17细胞的表面表型(CCR6+ CCR4+ CXCR3−)直接映射到功能表型(IL17表达),具有Th17.1表面表型的细胞(CCR6+ CCR4−CXCR3+)的功能不太明显,因为这识别了一种双重功能(IL17+IFNγ+)或单功能(IL17−IFNγ+) IFNγ表达细胞的混合细胞(图1和B.鲁斯等.[2],补充图E1)。结果来自在体外实验表明,双阳性(DP)细胞(CCR6 + CCR4 + CXCR3 +)是明显的双官能(IL17 +IFNγ+)[5.,可能代表了一个短暂的Th17细胞群,在包括IL12和肿瘤坏死因子(TNF)α在内的分化因子的存在下,这些细胞正在向功能性Th17.1表型过渡[6.]。可以说,作者之前已经显示了大多数效应T细胞的数据,其具有Th17.1表型的单型表达IFNγ,并且非常少(<5%)表达IL17 [5.]。
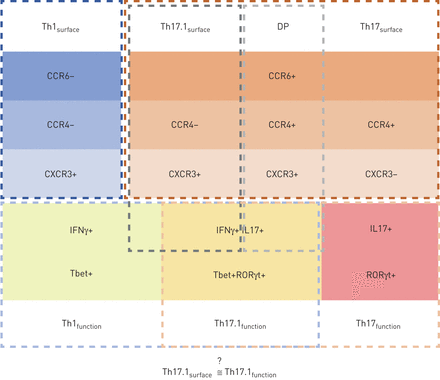
细胞统计学形式与功能:表面表达的趋化因子标记定义的T-helper (Th)1和Th17 t细胞亚群示意图(上半部分;蓝色和棕色柱)和由细胞内表达的功能标记物定义(下半部分;黄色、橙色和红色柱子)。DP:双阳性;干扰素:干扰素;IL:白介素。
kais等.[3.]和B.鲁斯等.[2然而,重申研究的主要T细胞表型本身归属于疾病的部位,其与功能性Th1-表型一致,并且两种研究都认为具有功能性Th17表型(单独表达IL17表达)的T细胞代表少数T细胞募集到肺的结节病。CCR6 + Th17细胞可以很适合于募集顺序病变,部分地通过包括CCL20的趋化因子的接合来募集到顺序病的肉芽肿炎炎症部位[7.]。大多数TH17细胞将通过局部细胞因子Milieu转化为非古典TH1细胞,其具有丰富的IL12和在rung病变中发现的TNFα。这些观察结果提供了支持使用抗TNF疗法进行结节病的理由,而且还提供了调查靶向TH17途径的治疗的原因[8.]。虽然关于Th17的精美知识与自身免疫相关联,但它是Th17细胞的可塑性,特别是它们在疾病部位转化为非古典TH1细胞的能力,这可能表明该促炎性T细胞的更广泛的作用粒状病症的肉芽肿疾病表型[9.那10.]。
最后,来自b的稿件鲁斯等.[2]有助于越来越多的知识破译Th17在结节病中的作用。ifn - γ的增强表达仍然是结节病的主要免疫学特征[11.]但是未来的研究是必要的,以确定IFNγ表达细胞的谱系(古典TH1细胞与非古典TH1 / TH17.1细胞)影响结节病的终点,包括赋予疾病致病性的差异(IE。保持肉芽肿炎症)[12.,作为临床状态的指标,预测对治疗的反应,以及在结节病中IL17的表达是否优于IFNγ作为疾病活动性的生物标志物。
脚注
支持声明:本工作由结节病研究,临床研究网络(S-07-007)和国家卫生研究院,全国心脏,肺和血液研究所(1R41HL129728)提供资金。本文的资金信息已存入Crossref资助者注册表.
利益冲突:没有宣布。
- 已收到2018年1月2日。
- 公认2018年1月20日。
- 版权所有©ers 2018











{kind=link}
{kind=link}