





文摘
对之间的关系具体呼吸道微生物群组成和毛细支气管炎的严重性。我们旨在识别鼻咽的微生物群简介和链接这些概要文件为毛细支气管炎急性严重性婴儿住院。
我们进行了一项多中心前瞻性队列研究的1005名婴儿(年龄< 1年)为毛细支气管炎住院三个冬天,2011 - 2014。通过应用16 s rRNA基因序列和聚类方法鼻咽吸入收集住院后24小时内,我们确定鼻咽的微生物群资料及其严重程度与毛细支气管炎。主要结果是重症监护使用,即。进入重症监护室或使用机械通气。
我们确定了四个鼻咽微生物群简介:三个配置文件之一嗜血杆菌,莫拉克斯氏菌属或链球菌,而第四最高细菌丰富。重症监护的速度是最高的婴儿使用嗜血杆菌占主导地位的概要,那些最低莫拉克斯氏菌属占主导地位的概要文件(20.2%与12.3%;未经调整的或1.81,95%可信区间1.07 - -3.11,p = 0.03)。调整后11患者的立场混杂因素,仍明显高于婴儿嗜血杆菌主要配置文件(或1.98,95%可信区间1.08 - -3.62,p = 0.03)。这些发现是在一个单独的外部验证为毛细支气管炎住院的307名孩子。
文摘
嗜血杆菌主要呼吸道微生物群同事与重症毛细支气管炎患儿的风险更高http://ow.ly/Bur6302vjRZ
介绍
毛细支气管炎是一个重要的全球公共卫生问题(1- - - - - -3]。事实上,细支气管炎住院的主要原因为我们婴儿,婴儿住院治疗上(占所有的18%3]。毛细支气管炎的严重程度的范围可以从一个小麻烦到致命的感染;然而,这种严重程度差异的原因还没有完全解释传统的危险因素,如年龄、早产和病毒病原学(4,5]。
而常见的病毒和细菌是识别使用传统技术(如。文化),这些病原体只代表一小部分微生物生活在任何一个人6,7]。基因组测序显示的最新进展的存在高度功能细菌社区居住人类,即。微生物群。新兴证据表明,呼吸道微生物群可以影响免疫反应(8- - - - - -13),这表明气道的作用微生物群在发展和发病率的急性呼吸道感染(阿里斯),包括毛细支气管炎(14]。然而,有限的文学协会的呼吸道微生物群与ARI发病率婴儿期是相互矛盾的。具体地说,阿里已经发现发病率更低(15和更高的16]婴儿更丰富莫拉克斯氏菌属属鼻咽。上述研究气道微生物群的外部验证他们的发现。此外,我们所知,没有研究调查呼吸道微生物群及其与疾病的严重程度的关系在婴儿毛细支气管炎住院;一个脆弱的人口发病率高。
在这种背景下,我们进行了一项多中心前瞻性群组研究毛细支气管炎的婴儿住院确定呼吸道微生物群简介和链接这些概要文件急性严重程度(即。重症监护使用和医院住院时间)。我们也在外部验证在一个单独的多中心研究结果为毛细支气管炎住院的儿童。
材料和方法
研究设计,设置和参与者
我们进行了一项多中心前瞻性队列研究的婴儿(年龄< 1年)为毛细支气管炎住院(重症毛细支气管炎)。这项研究名为35多中心气道研究协作(MARC-35) [17),是由急诊医学网络(EMNet,协调http://www.emnet-usa.org/),235年参与医院的合作。
使用一个标准化的协议,网站调查人员在美国14个州的17个站点(补充表E1)登记婴儿住院期间与毛细支气管炎的主治医生诊断连续三细支气管炎季节从11月1日至4月30日,2011 - 2014。毛细支气管炎是由美国儿科学会的指南定义为急性呼吸道疾病有鼻炎、咳嗽、tachypnoea、气喘、陶瓷器皿和弗雷德(18]。我们排除了婴儿与之前报名;那些被转移到参与医院>原住院后24小时;那些同意> 24 h后住院治疗;或与已知的心肺疾病、免疫缺陷、免疫抑制或胎龄< 32周。所有患者在医生的治疗。参与医院的机构审查委员会在每个批准了这项研究。家长或监护人的书面知情同意了。
数据收集
调查人员进行了结构化面试,评估患者的人口学特征、医疗和家庭历史,和急性疾病的细节。医院急诊科和图表评论提供进一步的临床数据,如生命体征、体检、医疗管理和处置。综述了所有数据EMNet协调中心和现场调查人员对缺失的数据查询和差异被手工数据检查。
鼻咽吸入物是由训练有素的网站收集的调查人员使用相同的标准化协议之前的队列研究中利用毛细支气管炎患儿的4,19]。所有网站使用相同的设备集合(美国Medline产业,Mundelein, IL)在24 h(住院和收集了样本。鼻咽样本添加到传输介质,立即放在冰,然后储存在−80°C。分批装运的冷冻样本干冰贝勒医学院,并检测1)17呼吸道病毒(如。呼吸道合胞病毒(RSV),鼻病毒)使用实时PCR检测(4,19,20.),2)使用16 s rRNA基因测序的微生物群。
16 s rRNA基因测序和组成分析
16 s rRNA基因测序的方法开发的方法是改编自美国国立卫生研究院(NIH)人类微生物组计划(6,7]。方法的细节描述的补充方法。短暂、细菌基因组DNA提取使用莫生物PowerSoil DNA隔离设备(莫生物实验室,卡尔斯巴德、钙、美国)。16 s rDNA V4地区扩大了PCR和测序MiSeq平台(美国Illumina公司,圣地亚哥,CA)使用一个2×250个基点paired-end协议收益率paired-end读取这几乎完全重叠。使用的引物扩增包含适配器MiSeq测序和单头条形码,允许池和PCR产物直接测序21,22]。
测序读双去复用基于独特的分子条形码和读取合并使用USEARCH v7.0.1090 [23]。稀疏曲线的细菌操作分类单位(辣子鸡)建立了使用每个样本序列数据,确保覆盖的细菌多样性。样品与次优的测序读re-sequenced确保大部分的细菌类群被包含在我们的分析。
16 s rRNA基因序列被聚集到辣子鸡97%的相似性截止值使用UPARSE算法(24]。辣子鸡是由映射质心到席尔瓦数据库(25)只包含16 s V4地区确定分类法。自定义脚本构建一个稀薄OTU表(稀疏了,只有一个序列深度)的输出文件中生成下游分析alpha-diversity(前两个步骤即。香农指数)和beta-diversity (即。加权UniFrac) [26,27]。我们利用多种质量控制措施,包括使用non-template控制微生物的DNA提取、16 s rRNA基因扩增,扩增子测序流程。质量控制措施的细节描述的补充方法。
结果测量
主要结果是重症监护使用,定义为进入重症监护室和/或使用机械通气(持续气道正压和/或插管在住院期间,无论位置)在任何时间在该指数住院(19,28]。二级结果医院住院时间≥3天,定义使用停留的平均时间为2天,类似于之前的研究中使用的方法(4,19]。
统计分析
对于每一个鼻咽样本,每个OTU的相对丰度计算。在属水平进行了分析;因为每个属是由一个OTU (如。检测到的嗜血杆菌属由单独的一个OTU),辣子鸡分配给相同的属都倒塌成一个小组报告(16]。识别鼻咽的微生物群资料,我们进行无偏集群通过分区medoids (PAM) [29日使用加权UniFrac距离)。每个PAM集群定义为一个点为中心(“medoid”)和最小化样本之间的距离在一个集群中。集群的数量选择的数据确定使用轮廓平均得分(补充图E1) (30.]。
此外,考虑到个体的存在和/或丰度属可能影响其他属的微生物群落,我们显示微生物群协会网络的基础上F的方法欧斯特et al。(31日]。方法的细节描述的补充方法。
接下来,在发现鼻咽癌微生物群概要文件,我们比较病人特点和医院使用卡方测试或者克鲁斯卡尔-沃利斯检验。检查协会的微生物群资料结果严重程度(即。重症监护的使用和医院住院时间≥3天),我们构建了两个逻辑回归模型的结果莫拉克斯氏菌属占主导地位的形象参考(15]。首先,我们只安装一个未经调整的模型,包括微生物群资料作为自变量。第二,我们构建了一个两级mixed-effects模型考虑到病人聚集在医院的水平。我们也调整为11个患者的立场变量(即。年龄、性别、种族/民族,妊娠年龄、呼吸问题的历史,托儿所出席,家里兄弟姐妹,一生中抗生素使用的历史,历史的皮质类固醇的使用,使用抗生素在pre-hospitalisation访问和呼吸道病毒检测PCR)。我们选择这些潜在混杂因素的临床合理性的基础上先天的知识(4,5,19,28,32]。我们没有调整标记的严重程度(如。生命体征),因为这些被认为是中间因素感兴趣的协会。
我们做了敏感性分析,以评估我们的研究结果的鲁棒性。首先,我们重复分析对病毒性病原体进行了统计处理,类似于之前的研究中使用的方法(4,33,34]。第二,我们模仿的长度保持计数变量通过拟合mixed-effects泊松回归模型。分析R v3.2的使用lme4mixed-effects模型和包phyloseq包(35]。假定值都是双尾,被认为具有统计显著性,p < 0.05。
结果
人口和序列
我们招收了1016名重症毛细支气管炎患儿从17家医院参与MARC-35。我们分析了婴儿的鼻咽样本所有16 s rRNA基因测序,获得17 399 260优质合并序列,其中16 685 637(95.9%)被映射到数据库。1016婴儿的样本,1005例(98.9%)有足够深度序列(每个样本稀疏截止:2128读),符合当前的分析。在分析群体,年龄中位数在住院3个月(四分位范围(差),2 - 6个月),60.0%是男性,42.6%为非西班牙裔白人。测序确定了24门379属。鼻咽微生物群是由三个属:链球菌(30.9%),莫拉克斯氏菌属(29.8%)和嗜血杆菌(20.2%);紧随其后的是普氏菌(2.3%)和葡萄球菌(2.2%)。
鼻咽的微生物群的概要文件和网络
PAM鼻咽微生物群的聚类识别四个截然不同的微生物群概况:1)嗜血杆菌占主导地位的概要文件(19.2%),2)莫拉克斯氏菌属占主导地位的概要文件(21.9%),3)链球菌占主导地位的概要(28.2%)和4)混合概要(30.7%)(图1)。
前三个配置文件是由之一嗜血杆菌,莫拉克斯氏菌属或链球菌属。相比之下,混合概要细菌丰富最高(p < 0.001)和alpha-diversity指数(香农指数(p < 0.001)的相对丰度最高普氏菌,Alloprevotella和韦永氏球菌属(补充表E2)。子集的混合配置文件确定三个类:子集co-dominated链球菌和莫拉克斯氏菌属,一个子集co-dominated链球菌和嗜血杆菌和一个子集的丰度相对较高链球菌,普氏菌和Alloprevotella(补充表E3)。
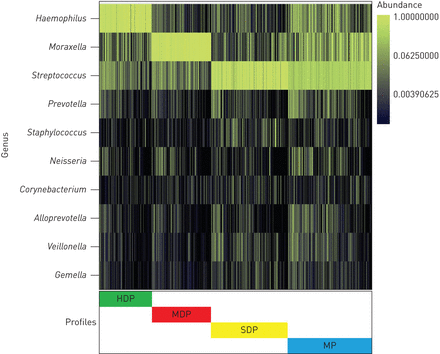
集群和1005年鼻咽微生物群组成婴儿毛细支气管炎住院,MARC-35。鼻咽微生物群资料的婴儿都是集群使用分区在medoids集群方法与加权UniFrac距离。彩色条显示四个微生物群配置文件:嗜血杆菌占主导地位的概要文件(黄芪丹参滴丸,绿色),莫拉克斯氏菌属占主导地位的概要文件(MDP,红色),链球菌,占主导地位的概要文件(SDP、黄色)和混合概要(MP,蓝色)。最优数量的集群被剪影指数。获得进一步信息的细菌组成样本内微生物群资料、十大最丰富的属现在显示在邻热图。分类描述在属水平因为每个属是由一个操作分类单位。
单个细菌的存在或丰度可能会影响他人的由于生态相互作用。检查微生物群的群落结构,我们创建了一个微生物群协会网络(补充图E2)。嗜血杆菌,莫拉克斯氏菌属和链球菌彼此属负相关,与观测一致嗜血杆菌占主导地位,莫拉克斯氏菌属占主导地位,链球菌主要配置文件。的链球菌属呈正相关韦永氏球菌属,也是积极联系在一起普氏菌和Alloprevotella。这个社区结构与混合微生物群概要文件是相一致的。
微生物群简介和病人的特点
病人的特点在四个不同微生物群概要文件(表1)。例如,婴儿的嗜血杆菌占主导地位的概要文件是旧的,不太可能非西班牙裔白人和更容易使用抗生素前指数住院婴儿相比与其他资料(所有p < 0.05)。在医院表示,婴儿嗜血杆菌占主导地位的形象也有更高的重量和更有可能接受抗生素在pre-hospitalisation访问(如。应急部门报名之前),唯一的鼻病毒感染(p < 0.001),而对婴儿和其他配置文件。相比之下,没有显著差异的生命体征。
微生物群简介和毛细支气管炎严重性
总的来说,161年(16.0%)婴儿住院所需细支气管炎重症监护使用。重症监护的速度是最高的婴儿使用嗜血杆菌占主导地位的概要,那些最低莫拉克斯氏菌属占主导地位的概要文件(20.2%与12.3%;未经调整的或1.81,95%可信区间1.07 - -3.11,p = 0.03;表2)。在多变量模型中调整11病人特征(如。人口统计,使用抗生素pre-hospitalisation访问期间,病毒)和集群在医院水平,利率仍明显高于与一个婴儿嗜血杆菌主要配置文件(或比较莫拉克斯氏菌属占主导地位的概要1.98,95%可信区间1.08 - -3.62,p = 0.03;表2和补充表E4)。也有积极的相对丰度之间的线性关系嗜血杆菌和重症监护的使用调整后的11个病人特征(p = 0.03;图2一个)。相反,在那些重症监护使用链球菌主导或混合配置文件没有明显不同于婴儿的参考莫拉克斯氏菌属占主导地位的概要文件。分层分析在病毒学地层E5补充表所示。尽管在大多数病毒地层的统计能力有限,与RSV感染和一个婴儿嗜血杆菌主导概要文件有更高的几率重症监护使用。此外,在另一个敏感性分析(n = 978),排除所有的婴儿没有病毒病原检测,结果并没有改变物质,即。重症监护的使用仍明显高于与一个婴儿嗜血杆菌主要配置文件(或比较莫拉克斯氏菌属占主导地位的概要2.15,95%可信区间1.16 - -3.99,p = 0.01)。
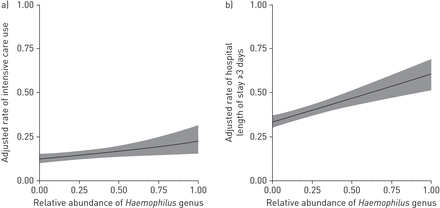
多变量关联的相对丰度嗜血杆菌属严重结果的速度在婴儿毛细支气管炎住院,MARC-35。二级mixed-effects模型构建集群占病人在医院的水平。模型调整为11个患者的立场变量(即。年龄、性别、种族/民族,妊娠年龄、呼吸问题的历史,托儿所出席,家里兄弟姐妹,一生中抗生素使用的历史,历史的皮质类固醇的使用,使用抗生素在pre-hospitalisation访问和呼吸道病毒检测PCR)。)有一个积极的线性的相对丰度之间的联系嗜血杆菌属和重症监护使用(调整后的速度0.1或1.07(每增加的相对丰度嗜血杆菌),95%可信区间1.01 - -1.13,p = 0.03)。b)有一个积极的线性的相对丰度之间的联系嗜血杆菌属,医院的住院时间≥3天(调整0.1或1.11(每增加的相对丰度嗜血杆菌),95%可信区间1.06 - -1.17,p < 0.001)。灰色阴影区域表示95%的置信区间。
主要结果的结果相似,与一个婴儿嗜血杆菌主导概要文件有较高的长度保持率≥3天比的莫拉克斯氏菌属占主导地位的概要文件(51.3%与33.2%;未经调整的或2.12,95%可信区间1.43 - -3.17,p < 0.001;表1和2)。在多变量模型中,仍明显高于与一个婴儿嗜血杆菌占主导地位的概要文件(或2.47,95%可信区间1.60 - -3.83,p < 0.001;表2和补充表E4)。也有积极的相对丰度之间的线性关系嗜血杆菌率和住院时间≥3天(p < 0.001;图2 b)。同样,在保持一个计数的灵敏度分析模型长度变量(即。二进制变量),微生物群之间的重要关联概要文件和结果保存(补充表E6)。
验证配置文件和与严重程度的关系
312年鼻咽样本选自儿童在MARC-30研究中,307名(98.4%)有足够的测序深度(每个样本稀疏截止:1064读)和用于验证。微生物群的PAM聚类方法的使用,使用的平均轮廓分数,显示四个微生物群概要文件(补充图E3)在MARC-35类似,即。三个档案由之一嗜血杆菌,莫拉克斯氏菌属或链球菌属,而第四个概要文件(即。混合)细菌的丰富性和alpha-diversity水平最高(补充表E7)。此外,混合配置的子集确定一个子集相对丰度高链球菌,普氏菌和Alloprevotella(补充表E8)。同样地,之间的重要关联嗜血杆菌占主导地位的概要文件和更高的严重结果也观察到,与更大的尺度效应,在这个嵌套病例对照样本更大程度的对比,例如,调整后的优势比为重症监护使用嗜血杆菌在与占主导地位的形象莫拉克斯氏菌属占主导地位的概要文件为5.34 (95% CI 1.96 - -14.5, p = 0.001;补充表E9)。
讨论
在这个前瞻性多中心的1005名婴儿重症毛细支气管炎,我们确定了四种不同的微生物群在鼻咽概要文件。婴儿用嗜血杆菌占主导地位的形象有显著较高的医院重症监护使用和长期呆比莫拉克斯氏菌属占主导地位的概要文件。相反,在那些结果链球菌主导或混合配置文件没有明显不同。这些发现是在一个单独的外部验证重症毛细支气管炎的多中心研究。据我们所知,这是第一个研究调查了协会的呼吸道微生物群与疾病严重程度在毛细支气管炎患儿严重。我们的数据证实并建立在先前的报道链接在气道细菌组成ARI结果(15,16,36- - - - - -39),两个研究的结果和临床重要性。
先前的报道在儿童急性呼吸道感染报道不一致的气道细菌和阿里的发病率和严重程度之间的关系。例如,Kloepferet al。(36]定量PCR应用于鼻样本(n = 380),发现之间没有关联流感嗜血杆菌检测与鼻病毒和阿里在学龄儿童严重程度。类似地,Carlssonet al。(37),使用COPSAC文化相关的技术2000年组(n = 283),没有发现任何关联流感嗜血杆菌下咽部和持续时间的检测在< 3岁儿童气喘发作。相比之下,Teoet al。,使用16 s rRNA基因测序在儿童哮喘研究队列(n = 234)在澳大利亚西部,报道6鼻咽微生物群的概要文件(嗜血杆菌,莫拉克斯氏菌属,链球菌,棒状杆菌属,Alloiococcus和葡萄球菌)在婴儿高特异反应性的风险,这一个嗜血杆菌占主导地位的鼻咽的微生物群与ARI发病率更高,更高的严重程度(16]。除了明显不一致嗜血杆菌阿里联系,我们也观察到大量的较低棒状杆菌属,Dolosigranulum和葡萄球菌在我们的人口属(即。婴幼儿毛细支气管炎住院)相比,在早期研究的健康儿童(15)和儿童有轻度上呼吸道感染(40,41]。潜在的在研究这些差异的原因包括患者群体的差异,临床设置(如。社区与住院病人设置),抽样(如。前鼻拭子与鼻咽吸入)和实验室技术(如。文化、PCR)细菌的鉴定。相比之下,我们的研究结果的有效性是通过使用16 s rRNA鼻咽癌基因测序的微生物群,样本容量大很多倍比其他任何研究这个话题之前,一个额外的多中心队列研究和验证的严重的细支气管炎。
观察到microbiota-severity协会挑战传统的以毛细支气管炎的视图。然而,这种微生物协会权证澄清的本质。可能有一个因果关系,即。的嗜血杆菌占主导地位的微生物群在婴儿的气道改变免疫反应增加毛细支气管炎的严重程度。事实上,Følsgaardet al。报道称,流感嗜血杆菌殖民的航空公司在无症状的新生儿与调节辅助(Th) 1 / Th2 / Th17-type上呼吸道粘膜的炎症反应(11]。Th2和Th17介质的存在表明,殖民流感嗜血杆菌可以抵消Th1反应需要根除病毒病原体的细支气管炎。同样,主支气管上皮细胞的研究表明,接触流感嗜血杆菌移植细胞间粘附molecule-1表达和增强后续感染RSV引起的趋化因子释放(42)和鼻病毒(42,43]。另外,一个嗜血杆菌占主导地位的微生物群可能仅仅是一个标记的婴儿容易发展严重细支气管炎。此外,反向因果关系,即。严重的疾病,导致快速生长嗜血杆菌在呼吸道,也可能44]。这些可能性并不是相互排斥的。尽管复杂,识别和验证的嗜血杆菌主导形象作为主要的罪魁祸首在呼吸道微生物群之间的联系和毛细支气管炎严重程度是一个重要的发现。我们的数据应该促进进一步的调查(如。动物模型,宏基因组,蛋白质组学,代谢组学)解开错综复杂的呼吸道微生物,病毒病原体、宿主免疫反应,和毛细支气管炎发病机理14]。
潜在的局限性
这项研究有几个潜在的局限性。首先,毛细支气管炎是一种疾病的航空公司,我们的研究是基于从婴儿的鼻咽样本。然而,降低气道抽样在婴儿技术和伦理挑战。然而,之前的研究已经表明强烈的上、下呼吸道微生物之间的相关性(45和病毒学46在孩子。因此,我们认为,婴儿的鼻咽微生物群可能表明下呼吸道。第二,使用16 s rRNA基因测序,我们没有分析微生物群在物种水平;因此,我们在确认有限的表型特征。第三,研究设计妨碍我们评估气道微生物生态系统的继承关系和呼吸道健康的孩子。为了解决这个重要的问题,研究人口目前正在跟随纵向6岁年与呼吸道标本在多个时间点取样。第四,对于任何观察性研究,微生物群的关联概要文件与毛细支气管炎不一定证明因果关系和严重程度可以通过无限的困惑的因素如病毒基因型和制度资源使用的变化。我们可以推测一个推论偏向由co-circulating病毒在医院或地区。然而,MARC-35登记病人在美国14个州17家医院在三个连续的细支气管炎季节(2011 - 2014)。他们的研究结果进一步验证了在一个单独的16-centre群2007 - 2010年期间严重细支气管炎。 Furthermore, the observed association between the microbiota and bronchiolitis severity remained significant after accounting for patient-level clustering within hospitals using a mixed-effects model. Fifth, we did not have information from a ‘control’ group (如。婴儿住院non-respiratory事件)。然而,这项研究的目的不是评估微生物群的作用对毛细支气管炎的发展(是/否),但调查其与疾病严重程度的关系。最后,尽管该研究人群包括种族,种族和地域多样化我们的样本严重细支气管炎,我们推断可能不是generalisable与动态设置。然而,我们的数据保持高度相关的130在美国每年000儿童住院;一个脆弱人群发病率高(3]。
结论
在这个前瞻性多中心队列1005婴儿毛细支气管炎住院,我们确定了四种不同的微生物群在鼻咽概要文件。婴儿用嗜血杆菌占主导地位的形象有更大的毛细支气管炎的严重性,即。明显较高的重症监护使用和长时间的滞留时间,比那些莫拉克斯氏菌属占主导地位的概要文件。这些研究结果验证了在一个单独的重症毛细支气管炎的多中心研究。这些发现应该作为一个重要的起点,进一步的机械和介入调查呼吸道微生物之间的相互作用,病毒病原体、宿主免疫反应和毛细支气管炎发病机理。
确认
我们感谢帕梅拉Luna莱斯大学(美国休斯顿,德克萨斯州)援助与统计分析。
下面的人MARC-35网站调查人员和合作者在本文。安妮·k·比斯利凤凰儿童医院,凤凰城,亚利桑那州,美国;胡安·c·青瓷,匹兹堡儿童医院,匹兹堡,PA,美国;阿里·r·科恩,马萨诸塞州综合医院,波士顿,MA,美国;米歇尔·b·邓恩,费城儿童医院的,费城,宾夕法尼亚州,美国;迈克尔•戈麦斯儿童医院在圣弗朗西斯,塔尔萨,好吧,美国;南希·英霍夫儿童医院在圣弗朗西斯,塔尔萨,好吧,美国;Sujit艾耶,戴尔儿童医疗中心的中央德克萨斯,奥斯汀,得克萨斯州,美国;费德里科•r . Laham阿诺德·帕尔默儿童医院,奥兰多,佛罗里达州,美国;查尔斯·g·Macias德克萨斯儿童医院,休斯顿,TX,美国; Thida Ong, Seattle Children's Hospital, Seattle, WA, US; Brian M. Pate, The Children's Mercy Hospital & Clinics, Kansas City, MO, USA; Wayne G. Schreffler, Massachusetts General Hospital, Boston, MA, USA; Michelle D. Stevenson, Kosair Children's Hospital, Louisville, KY, USA; Richard T. Strait, Cincinnati Children's Hospital and Medical Center, Cincinnati, OH, USA; Stephen J. Teach, Children's National Medical Center, Washington, DC, USA; Henry T. Puls, The Children's Mercy Hospital & Clinics, Kansas City, MO, US; Amy D. Thompson, Alfred I. duPont Hospital for Children, Wilmington, DE, USA; Vincent J. Wang, Children's Hospital of Los Angeles, Los Angeles, CA, USA; and Ilana Waynik, MD, Connecticut Children's Medical Center, Hartford, CT, USA.
脚注
可以从本文的补充材料www.qdcxjkg.com
支持声明:本研究支持的赠款U01 ai - 087881, R01 ai - 114552, R01 ai - 108588,和一下R21 hl - 129909来自美国国立卫生研究院(贝塞斯达,医学博士,美国)和威廉·f·弥尔顿基金的资助(美国波士顿)。资金信息,本文已沉积的资助者打开注册表。这个手稿的内容完全是作者的责任,并不一定代表美国国立卫生研究院的官方观点。
利益冲突:披露可以找到与这篇文章www.qdcxjkg.com
- 收到了2016年1月20日。
- 接受2016年7月18日。
- 版权©2016人队











{kind=link}
{kind=link}
![Multivariable association of relative abundance of Haemophilus genus with the rate of severity outcomes in infants hospitalised for bronchiolitis, MARC-35. Two-level mixed-effects models were constructed to account for patient clustering at the hospital level. The models adjusted for 11 patient-level variables (i.e. age, sex, race/ethnicity, gestational age, history of breathing problems, nursery attendance, siblings at home, lifetime history of antibiotic use, history of corticosteroid use, use of antibiotics during the pre-hospitalisation visit and respiratory viruses detected by PCR). a) There was a positive linear association between relative abundance of Haemophilus genus and the rate of intensive care use (adjusted OR 1.07 [per 0.1 increase in the relative abundance of Haemophilus], 95% CI 1.01–1.13, p=0.03). b) There was a positive linear association between relative abundance of Haemophilus genus and the rate of a hospital length of stay of ≥3 days (adjusted OR 1.11 [per 0.1 increase in the relative abundance of Haemophilus], 95% CI 1.06–1.17, p<0.001). The grey shaded areas represent the 95% confidence intervals.](http://www.qdcxjkg.com/content/erj/48/5/1329/F2.large.jpg?width=800&height=600&carousel=1){kind=link}
{kind=link}