





文摘
慢性阻塞性肺疾病(COPD)的标志是一个增强或炎症免疫反应异常肺部吸入粒子和气体,通常从香烟,特点是中性粒细胞的数量增加,激活巨噬细胞,激活淋巴细胞(Tc1和Th1细胞)。因此,抑制炎症反应是一个逻辑的慢性阻塞性肺病的治疗方法。尽管慢性阻塞性肺病的炎症性质,目前抗炎治疗在慢性阻塞性肺病患者和提供很少或根本没有好处可能有不利影响。出于这个原因,迫切需要发现有效且安全的抗炎治疗预防疾病的持续发展。近年来,注意力主要集中在招聘和抑制炎症细胞的活化,对立的他们的产品。在本文中,我们将一起总结的最先进的发展显然和/或潜在的有用的抗炎策略在慢性阻塞性肺病。
几个机械的概念与慢性阻塞性肺疾病(COPD)的发病机制(1]。慢性阻塞性肺病是一个增强的标志或炎症免疫反应异常肺部吸入粒子和气体,通常从香烟,特点是中性粒细胞的数量增加,激活巨噬细胞,激活淋巴细胞(Tc1和Th1细胞)。因此,抑制炎症反应是一种逻辑的方法治疗慢性阻塞性肺病的改善症状,比如咳嗽和粘液分泌,改善健康状况,减少发作。从长远来看,这种治疗方法应该减少疾病进展。然而,迄今为止,仍然没有有效的抗炎治疗也因为炎症慢性阻塞性肺病患者至少是部分glucocorticoid-resistant,可能因为吸烟和氧化应激损害组蛋白脱乙酰酶2 (HDAC2)函数(2]。在任何情况下,只有数量有限的药物已被证明影响的数量重要的炎症细胞,巨噬细胞,中性粒细胞和淋巴细胞),在COPD患者的肺。
很明显,有一个巨大的未满足的医疗需要关于有效抗炎剂治疗慢性阻塞性肺病患者。出于这个原因,注意力主要集中在抑制招聘和激活这些细胞,对抗他们的产品,虽然它并不少见的许多类药物同时执行这两个操作。此外,还有药物可能间接的抗炎作用。
药物直接影响炎症的细胞成分
目前,一些新的药物直接抑制炎症的细胞成分,尽管人们担心他们的使用,因为受损的中性反应可以增加对感染的易感性在COPD患者中,他们通常已经在风险(3]。治疗探索包括磷酸二酯酶4 (PDE4)抑制剂,腺苷2受体受体激动剂和药物干扰粘附分子(表1)。
PDE4抑制剂
PDE4的同工酶是一个主要的治疗目标,因为它是主要的同工酶在大多数炎症细胞参与COPD的发病机制。其抑制炎症细胞的影响几个特定的反应,如生产和/或释放促炎介质,包括细胞因子和活性氧物种(4),慢性阻塞性肺病的证据确凿的效果在动物模型(5]。在气管平滑肌PDE4也存在,但到目前为止选择性PDE4抑制剂没有显示急性支气管扩张剂人类活动(6,7]。
大多数的慢性阻塞性肺病(长期)研究迄今为止进行的第二代口服PDE4-inhibitors, cilomilast和roflumilast8]。cilomilast终止,因为大型多中心发展的第三阶段试验未能达到他们的预定义的功效端点和经常与胃肠道副作用。相反,roflumilast已经在欧洲多个国家注册,在美国批准。
几个6 - 12个月的随机临床试验(相关的)表明,roflumilast提供持续的临床疗效,主要减少发作,在慢性阻塞性肺病患者的一个子集的特征包括慢性支气管炎并发吸入糖皮质激素(有/没有9]。重要的是,它减少慢性阻塞性肺病急性加重和改善肺功能(pre -和post-bronchodilator用力呼气量在1 s (FEV1与长效β)尽管伴随治疗2受体激动剂(10]。汇集的数据分析表明,副作用与roflumilast(恶心、头痛、腹泻和体重减轻)通常是不严重和自限性,虽然导致增加病人退出整个研究[8]。
其他几个口语PDE4抑制剂仍在发展表1),尽管到目前为止,口服PDE4抑制剂的治疗作用是有限的副作用,特别是呕吐和恶心(表1)[6,8]。
有人建议,这些副作用是由于特定亚型PDE4的抑制,PDE4D [11),而PDE4B比PDE4D更重要在炎症细胞(12]。理论上,PDE4B-selective抑制剂可能有一个更大的治疗比(8]。然而,这个观点不是普遍共享和最近发现的选择性brain-penetrant PDE4抑制剂,没有呕吐(13)进一步增加了对避免PDE4D的目标体重,尤其是当这种酶亚型表达在细胞感兴趣的慢性阻塞性肺病(8]。
另外,PDE4抑制剂口服生物利用度较低可能是由吸入其功效最大化治疗炎性疾病而最小化其副作用。几个吸入PDE4抑制剂,例如GSK256066,正在开发,但是失败在临床试验中已经报道了另外三个吸入PDE4抑制剂,tofimilast [14],awd12 - 281 [15]和UK500001 [16]。
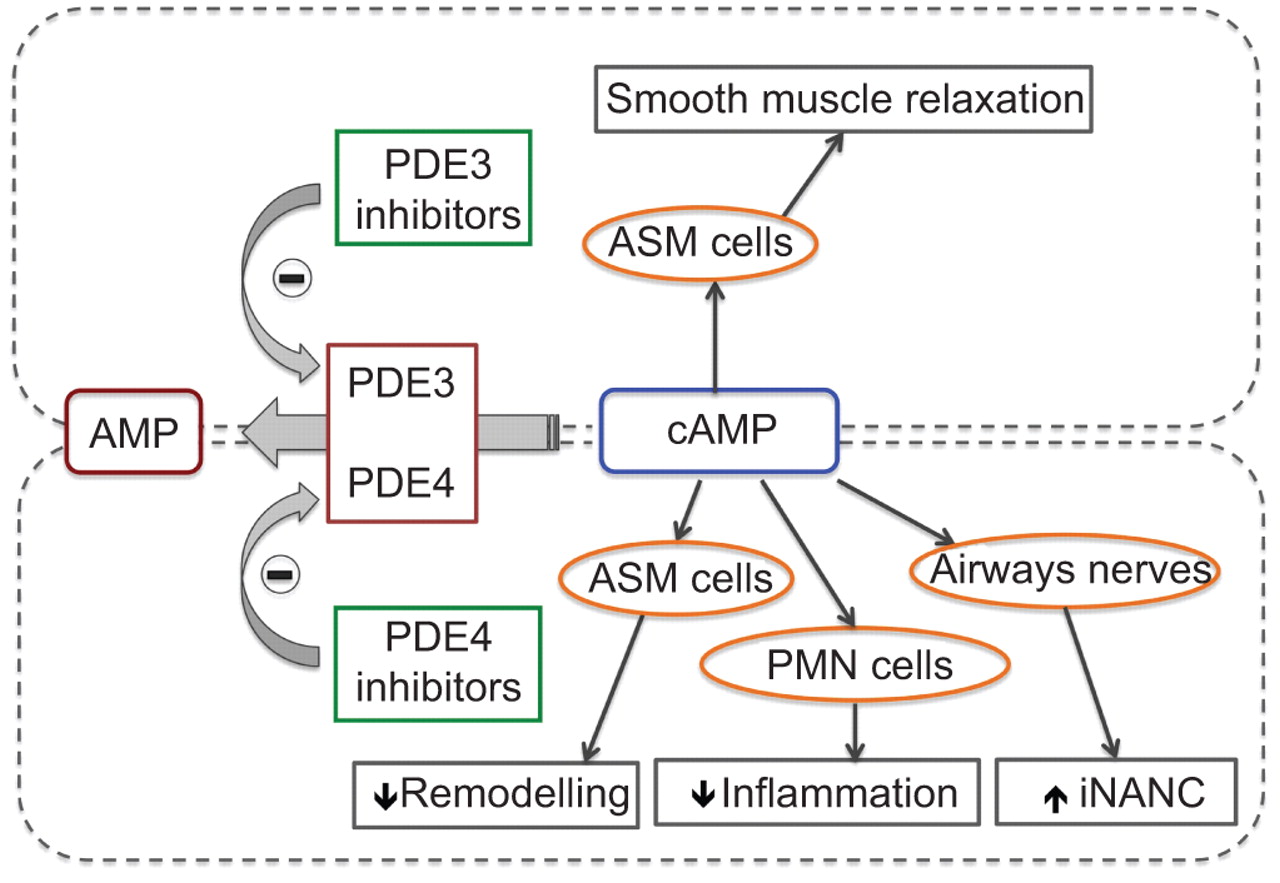
另一种方法是混合PDE3/4抑制剂的发展的目标有支气管扩张剂和抗炎活动在一个分子(图1)。混合PDE3/4抑制剂的发展与长期的行动加上抗炎活动可能在慢性阻塞性肺病更大的效用,因为两者的结合支气管扩张剂(PDE3-mediated)和抗炎活性(PDE3 -和PDE4-mediated)可能导致一个增强的总体疗效相比选择性PDE4抑制剂。RPL 554是最先进的混合PDE3/4抑制剂的发展(17]。
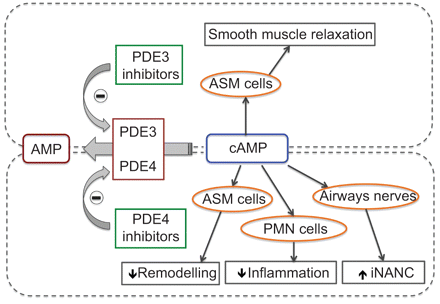
结合抑制磷酸二酯酶(PDE) 3和PDE4抑制剂有添加剂和协同抗炎和bronchodilatory效果与抑制PDE3或PDE4的孤独。背后的机制(s)的明显的协同效应双重PDE3/4抑制作用尚不清楚。然而,有人认为,PDE3(主要是局部颗粒细胞分数)和PDE4(主要是胞质)可以调节不同池的阵营。ASM:气道平滑肌;中性粒细胞:多形核中性粒细胞;iNANC:抑制non-adrenergic non-cholinergic。
此外,混合PDE4/7抑制剂正在发展中。与PDE4抑制PDE7抑制可能的结合,在理论上,是协同减少炎性细胞激活和肺部趋化因子和细胞因子释放8]。TPI 1100是由两个反义寡核苷酸瞄准的mRNA PDE4B / 4 d和PDE 7一个亚型18]。很有效地降低中性粒细胞涌入和关键细胞因子在慢性阻塞性肺病的吸烟小鼠模型建立(18]。
腺苷一2受体受体激动剂
有证据表明,腺苷酸受体参与炎症过程。到目前为止,四个亚型(A1,一个2,一个2 b和一个3腺苷酸受体的克隆。腺苷酸的抗炎效应通常归因于入住率的环磷酸腺苷(营)提高G年代-protein-coupled一2受体(19]。一个关键分子机制的抑制核转录因子(NF) -κB通路激活细胞因子如肿瘤坏死因子(TNF) -α和白介素(IL) 1β。的刺激2巨噬细胞炎性细胞因子受体限制生产,减少对内皮细胞粘附分子的表达,抑制超氧化物阴离子的生成和白三烯的合成中性粒细胞(20.- - - - - -22]。
几个腺苷2受体受体激动剂已报告在COPD模型是有效的(表1),但潜在问题可能导致心血管副作用,主要是低血压和反射性心动过速20.,22]。Regadenoson (cvt - 3146),选择性腺苷2受体激动剂,安全的妥协是门诊病人临床稳定的中度至重度慢性阻塞性肺病,但无法修改多个肺功能参数,包括重复FEV1与用力肺活量(FVC)相比安慰剂(23]。然而,UK432,097,有利的犀牛豚鼠肺部没有任何明显的心血管副作用,可能GW328267X,是高度有效的2受体激动剂,目前在慢性阻塞性肺病的二期试验。报告的影响这些化合物在慢性阻塞性肺病饶有兴趣地等待。
最近,它提出了发展2受体受体激动剂合成高活性化合物的活性,磷酸化形式2受体受体激动剂(24,25]。这种方法可以帮助实现所需的抗炎行动和减少不必要的副作用,如低血压。
药物干扰粘附分子
炎症过程在慢性阻塞性肺病的特点是一个持续的炎症细胞的迁移,主要是中性粒细胞,从血管腔室到肺,这部分是由selectins [26]。Selectins调解瞬时胶粘剂互动与炎症相关的识别碳水化合物抗原决定基,sialyl刘易斯x(系统性红斑狼疮x),表示结构多样化protein-lipid配体在循环白细胞。快速周转selectin-ligand债券,由于他们在一起,把利率非常高强度的优势,使他们能够调解细胞拘束和滚动在剪切流27]。三个已确定selectins: L - P -和E-selectin。
假设,针对这些分子可能会减少炎症的慢性阻塞性肺病(28]。绝对最常见的方法在抑制selectin函数是通过直接抑制selectins的一个或多个。碳水化合物,重组可溶性配体、抗体和小分子抑制剂都进入临床开发潜在的治疗药物针对selectins (表1)。
Bimosiamose (TBC1269, 1)是合成目标E - pan-selectin拮抗剂,P -, L-selectin。在体外bimosiamose块粘连的中性粒细胞,嗜酸性粒细胞和淋巴细胞在静态和动态流条件下29日]。在活的有机体内,它显示抗炎功效在各种动物模型,包括肺部炎症模型(30.]。在试点试验,吸入bimosiamose安全和耐受性良好稳定的轻度至中度COPD患者,鼓励在痰液抗炎作用参数,减少引发水平和淋巴细胞(31日]。bimosiamose的另一个最近的试验证明,吸入支气管扩张剂28天的标准是77年安全和耐受性良好中度到重度COPD患者(慢性阻塞性肺疾病的全球倡议阶段ii iii) (32]。它导致了广泛而显著的衰减的气道炎症和肺功能改善的趋势。显然,还需要进一步的研究来证明一个真正的临床效益bimosiamose COPD患者。
肝素是一种已知的抑制剂的selectin-mediated交互。pgx - 100 (2 o, 3点desulfated肝素)和pgx - 200, pgx - 100的吸入配方,开发最大化anti-selectin肝素的活性,同时尽可能减少抗凝血效果,但IIb阶段pgx - 100试验在慢性阻塞性肺病急性加重患者终止是由于临时分析的结果显示安全性没有有效性的证据(33]。
EL246, anti-selectin单克隆抗体(mAb)绑定到一个特定的抗原决定簇E -和L-selectins和抑制细胞粘附功能,在会展治疗慢性阻塞性肺病急性加重(33]。
抑制剂的炎症介质
细胞因子和趋化因子调节一系列炎症细胞的迁移和活化在慢性阻塞性肺病和现在有一个密集的搜索化合物能够与这些炎症介质。抗炎介质或刺激生产管理被认为是一个有趣的促炎和抗炎治疗的可能性,因为失衡可能构成慢性肺部炎症介质(3]。
治疗炎症介质的影响目前正在调查:TNF-α抑制剂,趋化因子抑制剂,NF-κB抑制剂,p38增殖蛋白激酶(MAPK)抑制剂,磷酸肌醇3-kinase (PI3K)抑制剂,白三烯(LT) B4抑制剂、过氧物酶体proliferator-activated受体(PPARs)受体激动剂,大环内酯类和他汀类药物(表2)。
TNF-α抑制剂
TNF-α被认为发挥核心作用在慢性阻塞性肺病的病理生理学34]。t细胞是由肺泡巨噬细胞、中性粒细胞、肥大细胞和上皮细胞接触不同的污染物,包括吸烟。在动物模型中,TNF-α诱发病态特性与慢性阻塞性肺病联系在一起,如炎症细胞渗透到肺,肺纤维化和肺气肿。此外,它能增强中性粒细胞趋化性迁移,诱导趋化因子的表达CXCL8(也称为引发)和移植内皮粘附分子。在活的有机体内,高浓度的TNF-α已经证明在外周血、支气管活检、诱导痰、支气管肺泡灌洗液的稳定的慢性阻塞性肺病患者与对照组相比。一起TNF-αIL-1β已被确定为一个关键细胞因子能够启动在慢性阻塞性肺病加重病人的炎症级联。特别是,据报道,TNF-α是初始和预测细胞因子释放的级联后暴露在脂多糖(LPS)。因此毫不奇怪,有人建议,阻断TNF-α的生物效应可能在慢性阻塞性肺病的治疗是有益的。
有三个商用生物制剂抑制TNF-α:服用依那西普、英夫利昔单抗和adalimumab。此外,其他两个肿瘤坏死因子抑制剂,certolizumab pegol golimumab,都在发展。他们是有效的治疗炎症性疾病,如风湿性关节炎和炎症性肠病,和他们的使用可以延长慢性阻塞性肺病。然而,随机对照试验评估的有效性TNF-α抑制剂在COPD患者数与第一个研究的结果不是非常有前途的(35- - - - - -37]。此外,infliximab-treated主题和肺炎的发生率较高,虽然不具有统计学意义,更多的癌症病例被报道(36]。
尽管如此,一项观察性研究进行了评估的有效性TNF-α拮抗剂在预防慢性阻塞性肺病住院治疗上一群病人诊断为风湿性关节炎和慢性阻塞性肺病识别健康声明数据库,表明TNF-α抑制剂与减少有关COPD住院率对COPD患者接受这些药物来治疗类风湿性关节炎(38]。然而,这种效果是由于只减少50%的慢性阻塞性肺病住院服用依那西普,一个完整的人组成的二聚的融合蛋白TNF-αII型受体和Fc免疫球蛋白G1的一部分。其他TNF-α抑制剂的研究,即英夫利昔单抗,不降低COPD住院的风险。
必须强调,TNF-α的浓度在慢性阻塞性肺病患者的血液或肺部有很大差异。血清浓度的TNF-α慢性阻塞性肺病患者在24周英夫利昔单抗研究不是特别高36),所以可能是这种疗法是有效的在慢性阻塞性肺病患者的一组中,TNF-α更丰富。我们也假设TNF-α扮演一个重要角色在疾病早期,几乎不太可能在更高级的疾病。英夫利昔单抗研究的大多数患者中度到重度COPD患者(36),这也可能是为什么没有回应治疗。
在任何情况下,TNF-α以两种形式存在,膜结合proform由233个氨基酸分子质量的26 kDa, 17 kDa的可溶性形式由157 nonglycosylated氨基酸。已经表明,生物活性的分解TNF-α从membrane-anchored proform是由一种金属蛋白酶叫TNF-α转换酶肝动脉化疗栓塞(TACE)和抑制块释放TNF-α[3]。在气道炎症动物模型,PKF 242 - 484和PKF 241 - 466,两个TACE抑制剂,阻塞TNF-α释放到航空公司和炎性细胞涌入3]。到目前为止还没有TACE抑制剂达到市场。这是部分原因别说话的一般缺乏选择性抑制剂(3]。
治疗患者TNF-α生产的抑制剂,或与反义寡核苷酸针对mRNA分子编码TNF-α可能替代策略探索(3]。
趋化因子抑制剂
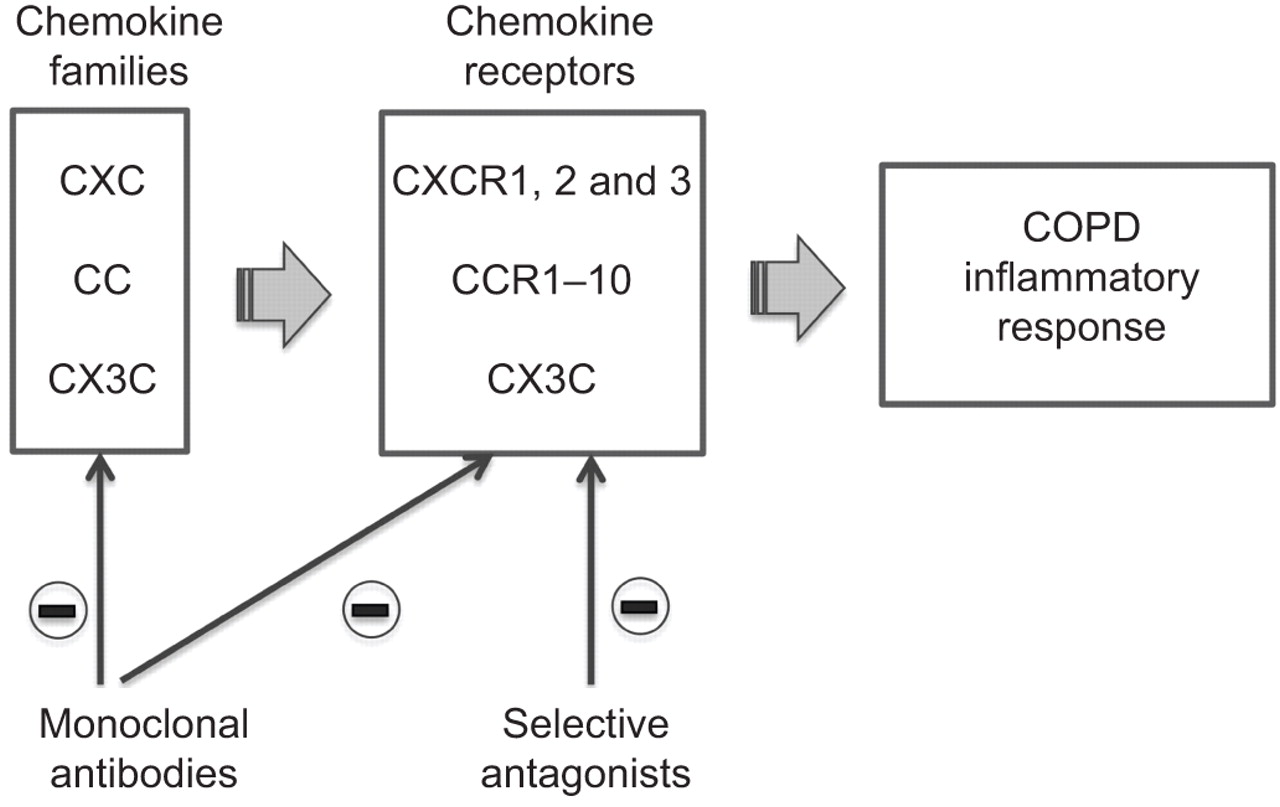
炎症趋化因子在感染或组织损伤产生各种各样的细胞,包括居民和白细胞渗透,以应对产品细菌和毒素或炎性细胞因子il - 1、TNF-α和干扰素。调节增加肺部炎症细胞迁移和活化。趋化因子分为四个主要类别的基础上的数量和间距保守的半胱氨酸的氨基酸:科学家、CC、C和CX3C家庭。他们通过G-protein-coupled受体信号。
ABX-CXCL8 anti-CXCL8马伯,在慢性阻塞性肺病的II期临床试验,测试显示改善过渡呼吸困难指数,但没有肺功能改善或治疗病人的健康状况39]。ABX-CXCL8只有承认自由趋化因子(40),但趋化因子的活性形式是必然内皮表面蛋白聚糖(41]。这可能是一个可能的解释这种抗体的临床失败。
也为了克服这个问题,有人建议治疗目标通过阻断趋化因子系统中的交互与抗体或小分子抑制剂防止招聘和激活引起的白细胞趋化因子(图2)[42]。然而,一个潜在挑战将军在慢性阻塞性肺病是针对趋化因子趋化因子的冗余网络这样一个受体或抑制趋化因子可能不足以阻止炎症反应(43]。
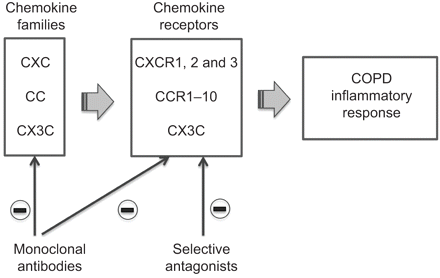
趋化因子和趋化因子受体的示意图表示潜在目标为新药物对慢性阻塞性肺病(COPD)患者。
随着几个趋化因子可以激活一个受体,其作用是最好的讨论通过其受体,这对科学家分为受体趋化因子(CXCRs)和CC趋化因子(ccr自己)(表2)[44]。
CXCL8激活中性粒细胞通过一个特定的受体(CXCR1)耦合的激活和脱粒,和通过高亲和性受体(CXCR2),在趋化性是很重要的。COPD动物模型,封锁CXCR1和CXCR2由特定抑制剂显著降低中性气道炎症(45]。CXCR2拮抗剂可能更有用,因为CXCR2在单核细胞也表达了。一些CXCR2拮抗剂正在发展的潜在治疗慢性阻塞性肺病(表2)。在健康志愿者(SCH527126抑制臭氧感生sputum-derived嗜中性46),和某人- 656933阻塞agonist-induced upregulation CD11b外周血中性粒细胞的健康受试者,臭氧感生的作用与抑制嗜中性(47]。这些发现表明CXCR2拮抗剂治疗COPD患者的潜力。尽管如此,对许多行动的影响CXCL8和相关趋化因子作用于CXCR2必须严密监控为中性粒细胞对宿主防御病原微生物是必不可少的,和不受欢迎的免疫抑制无疑是最令人担忧的潜在不利影响管理这些化合物对人类的48]。
CXCR3,另一种趋化因子受体的表达是提升航空公司外围的吸烟者和COPD患者49),是一个潜在的目标为小分子拮抗剂或抗体。CXCR3的封锁应该防止炎症细胞达到炎症,从而缓解疾病的网站。几个小分子拮抗剂CXCR3已报告(表2)。
CX3CL1,存在膜结合蛋白和可溶性趋化因子,可以调节白细胞粘附和函数是一种有效的化学引诱物(50]。它只是绑定到,独特的配位体CX3C趋化因子受体1 (CX3CR1)。吸烟引起的肺气肿的小鼠模型,CX3CR1 +细胞进入肺的涌入(51]。此外,CX3CL1表达式是调节在吸烟者的肺组织与慢性阻塞性肺病52]。这使得这一对中的一个有吸引力的治疗目标53]。抗体或小分子可以用来阻止CX3CL1-CX3CR1交互(表2)。他们可能减少或防止白细胞渗透/积累、结构改造/破坏和功能下降的开发和进展慢性阻塞性肺病(54]。然而,CX3CR1拮抗剂治疗发展必须追究谨慎因为CX3CL1有许多重要的生理作用,包括诱导抗肿瘤活性和保护神经退行性疾病(54]。
行动的CC-chemokines CCR2-receptor也已被证明能够参与慢性阻塞性肺病(54]。临床前研究表明CCR2扮演重要的角色在单核细胞和巨噬细胞对网站流量的炎症。几个选择CCR2受体拮抗剂和anti-CCR2马伯(mln - 1202)正在调查(表2);所有证明有前途的临床活动(54,55]。然而,CCR2序列同源性低和配体之间的人类和物种意味着低的一个关键问题是阻碍CCR2能否产生足够的临床疗效在COPD患者56]。
NF-κB抑制剂
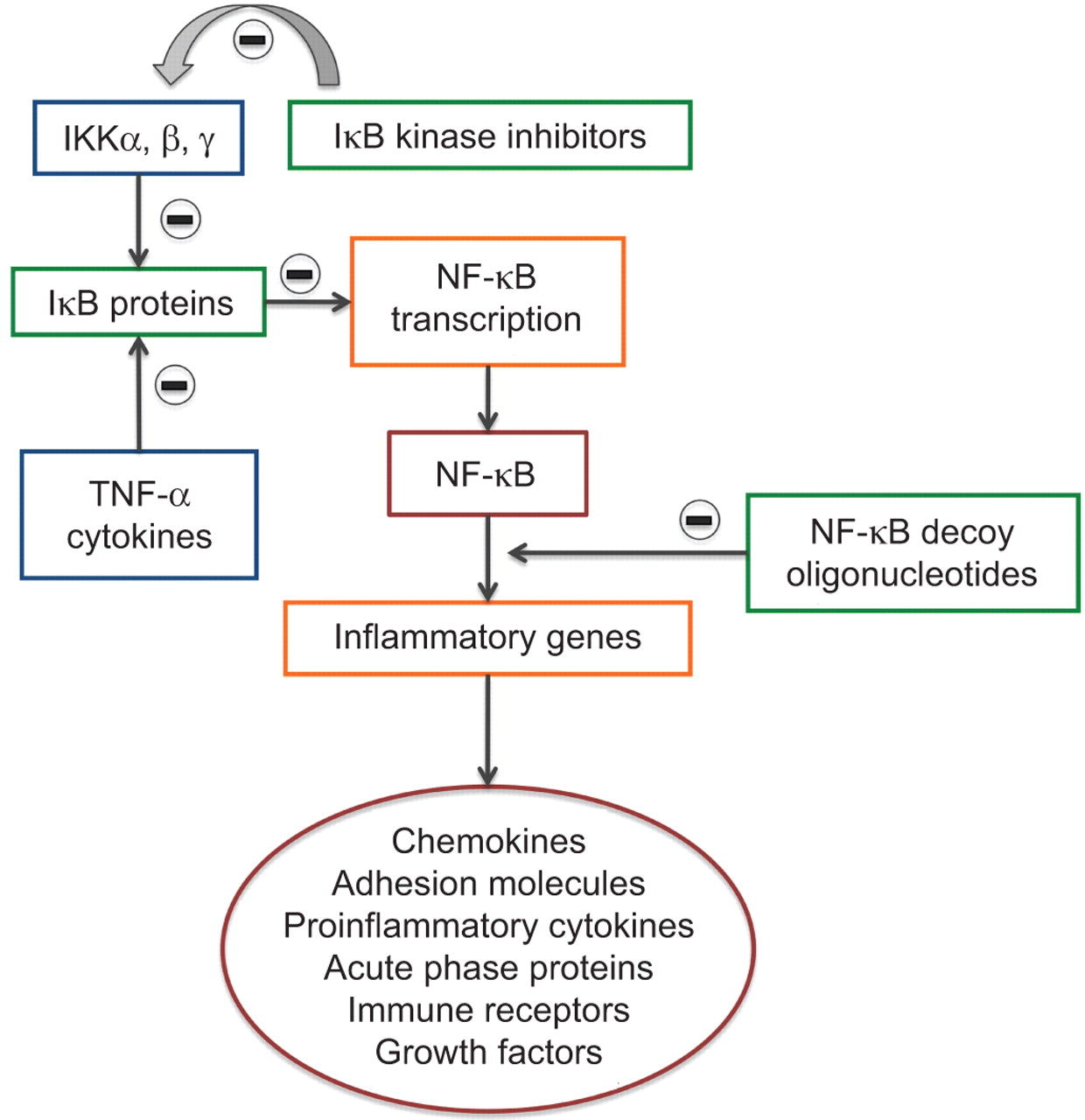
大量的证据令人信服地证明了一个明确的角色NF-κB转录因子及其信号激酶在稳定和恶化形式的慢性阻塞性肺病(图3)[57]。NF-κB抑制剂(IκB)家族的蛋白质调节NF-κB-dependent通过抑制DNA结合转录和细胞细胞质本土化这些因素。IκB IκB蛋白质磷酸化的激酶组成的复杂的至少三个蛋白质,κB激酶抑制剂(IKK)α,IKKβIKKγ。IKKα和IKKβ是两个催化亚基,而IKKγ调节亚基(NF-kB基本调制器;NEMO)。基因打靶实验显示所需的许多炎性刺激IKKβ单元NF-kB激活(58),而IKKα只扮演一个角色响应某些刺激,在有限数量的细胞类型。
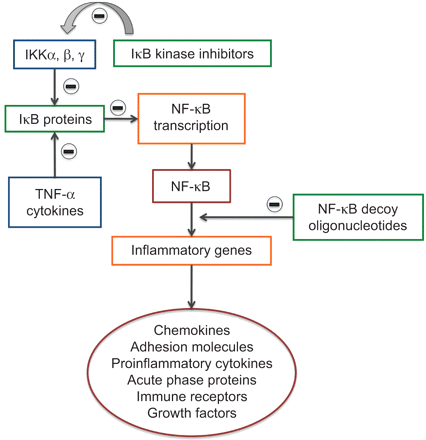
规范和非规范通路核因子(NF) kb。IKK:κB激酶抑制剂;NF-κB IκB:抑制剂;肿瘤坏死因子:肿瘤坏死因子。
有几个可能的NF-κB的抑制方法。他们包括IκB基因转移、IKK NF-κB-inducing激酶和IκB泛素连接酶,调节NF-κB和毒品的活动,抑制IκB[的退化59]。这些药物正在研制的几个(表2)[60]。bms - 345541是一个高度的选择性抑制剂IKK具有良好的药代动力学特征(口服生物利用度100%,静脉半衰期2.2小时),il - 6的表达抑制TNF-α-induced引发和eotaxin剂量依赖性的气道平滑肌细胞(61年]。在人类气管平滑肌细胞,co-incubation bms - 345541显著抑制了易位NF-kB TNF-α引起的核和IL-13 [62年]。ps - 1145能够诱导剂量依赖性抑制磷酸化IkBα和NF-κB激活,因此,它能减少炎症因子的表达包括粘附分子、细胞因子和趋化因子对气管平滑肌细胞(63年]。
进一步发展包括NF-κB“诱饵”寡核苷酸(64年)和反义和小核RNA)代理(57]。“诱饵”寡核苷酸,双链DNA寡核苷酸被用来作为转录因子的诱饵,竞争性结合自由NF-κB二聚体从而防止cis-acting网站启动子区域内的相互影响。反义药物使用稳定phosphothionate寡核苷酸结合互补的信使rna,从而阻碍翻译。siRNA代理是基于核酸的目标NF-κB的信使rna通过RNA干扰,减少其丰富的过程。
NF-κB担心长期抑制之一,然而,是有效的抑制剂可能会导致免疫抑制和削弱主机防御。然而,有NF-κB的替代途径激活,通过激酶IKK以外,这可能是更重要的在调节炎性疾病(28]。
p38 MAPK抑制剂
MAPKs属于丝氨酸/苏氨酸激酶家族能传感环境刺激细胞核和控制细胞周期机械、细胞死亡,基因转录和蛋白质翻译。他们在慢性炎症中发挥关键作用。MAPKs在一连串的顺序激活蛋白激酶和细分为三个主要途径:细胞外signal-regulated激酶(ERK) c-Jun n端激酶(物)2,p38激酶(65年]。ERK通路刺激尤其是通过g蛋白耦合和生长因子受体参与细胞增殖、分化和存活,而JNK2和p38激活主要由细胞因子与炎症和细胞凋亡。
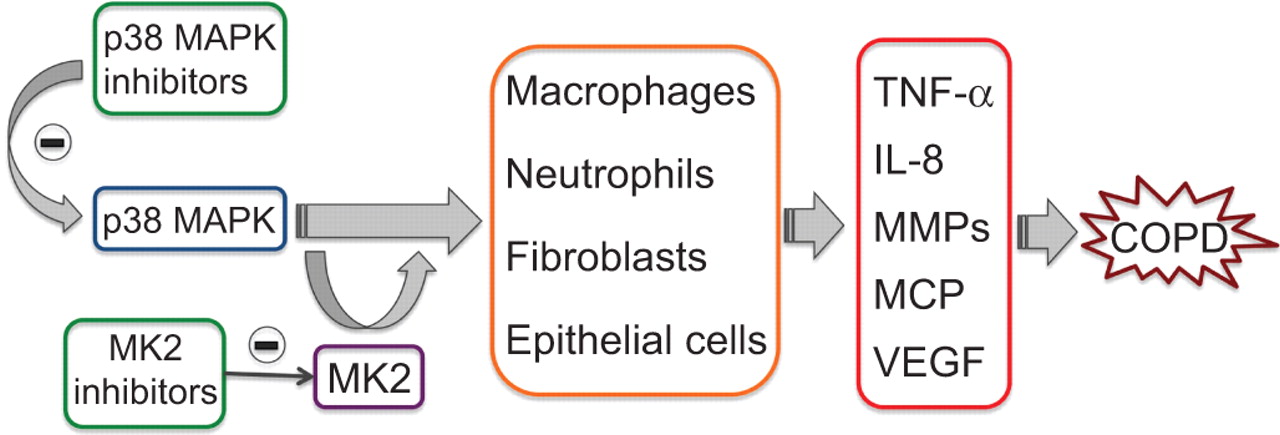
p38 MAPK似乎扮演一个重要角色在慢性阻塞性肺病(图4)[66年]。其激活炎性细胞类型与疾病在关键起始和进展67年]。p38 MAPK似乎也是最有效的MAPK在稳定,在转录后水平,相关的细胞因子和趋化因子的mrna COPD发病机理(68年]。p38 MAPK的四种不同的亚型,指定的α、β、γ和δ。p38α发现白细胞主要同种型,上皮细胞,平滑肌细胞,而p38δ是更多的巨噬细胞中高度表达和p38γ骨骼肌(69年]。
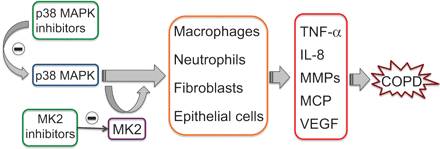
p38增殖作用的蛋白激酶(MAPK)是表示大多数炎症细胞和调节生产重要的炎症介质。不仅抑制p38 MAPK有望抑制促炎细胞因子的生产还他们的行为,从而打断了恶性循环,通常发生在慢性阻塞性肺疾病(COPD)。MK2型,p38 MAPK的下游底物,代表另一个优秀的抗炎治疗的目标。肿瘤坏死因子:肿瘤坏死因子;IL:白介素;MMP:基质金属蛋白酶;MCP:单核细胞趋化蛋白;VEGF:血管内皮生长因子。
抑制p38 MAPK因此关注作为治疗目标发展的新型抗炎剂治疗慢性阻塞性肺病(表2)[70年,71年]。抑制剂结合的竞争力在磷酸腺苷的口袋和目标p38α和β但不是δ或γ,可能提供一个广泛的抗炎作用,因为α异构体中含量最丰富的同种型炎性细胞。他们在改善显示效果几个COPD-like病态在动物模型72年]。
在有限合伙人和博来霉素模型分别炎症和肺纤维化,某人239063年减少中性粒细胞浸润,il - 6表达,基质金属蛋白酶9 (MMP)航空公司的活动(72年]。另一个p38 MAPK抑制剂,doramapimod (BIRB 796)是为了诱导剂量依赖性抑制在LPS-induced TNF-α生产在人类主题,实现97%至88抑制在50和600毫克的剂量,分别为(73年),但dose-limiting肝毒性预防使用大剂量(74年]。Doramapimod能够抑制α、β和γ亚型,并抑制几个non-p38激酶(71年]。
Dilmapimod,抑制p-HSP27 TNF-α生产从全血从COPD患者(75年),和PH797804减少气道和系统性炎症反应引起的有限合伙人吸入在健康受试者76年),已经在慢性阻塞性肺病患者进行测试。四周治疗dilmapimod引起痰中性粒细胞减少和血清纤维蛋白原,但不是在血清c反应蛋白,引发,IL-1β或il - 6水平;这是伴随着FVC但FEV的改善1(77年]。六周治疗PH797804诱导FEV的显著增加1伴随呼吸困难评分的改善,吸气量和后血清CRP水平持续减少每日口服一定剂量的3、6和10毫克·天−16周(78年]。
然而,不良事件的可能性造成不希望的药理作用是主要关心p38抑制剂类药物(79年]。此外,有担心长期使用p38 MAPK抑制剂因为这些抑制剂是有效抑制先天免疫反应的病毒和细菌感染(80年]。
为了减少不必要的系统性副作用的风险,有人建议,p38 MAPK抑制剂应该直接发送到肺(70年,81年]。ARRY371797和PF03715455 p38 MAPK的有力和高度选择性抑制剂在体外和在活的有机体内。不过,他们尚未在COPD患者进行测试。也p38 MAPK反义寡核苷酸的方法可能有慢性阻塞性肺病的治疗潜力,但从来没有在慢性阻塞性肺病(3]。
必须强调,p38αMAPK可能不是最优的目标发展的抗炎药,因为它参与反馈控制回路,抑制“上游”的活动MAPK激酶激酶(MAP3Ks),如转变增长factor-activated激酶1 (TAK1)和mixed-lineage激酶(灵魂)2 / MLK3中涉及的其他促炎通路的激活,如导致JNK2的激活和IKKβ[82年]。药物抑制p38αMAPK取消这些反馈控制回路,导致TAK1 hyperactivation和灵魂,因此JNK2 hyperactivation。
MK2型抑制剂
MAPK-activated蛋白激酶2 (MK2型),p38 MAPK的下游底物,是一个更具体的目标,作用于有限的下游底物(图4),从而最小化参与各种介质。此外,它在炎症的发展中扮演多个角色及其抑制预计将产生相同的有益影响p38 MAPK抑制副作用较小(83年]。尽管MK2型抑制是一种很有前途的目标,提出了许多问题,因为它是表达各种各样的细胞和控制大量的功能。MK2型也可以激活信号级联除了p38途径及其抑制可能产生一些不受欢迎的副作用83年]。
PI3K抑制剂
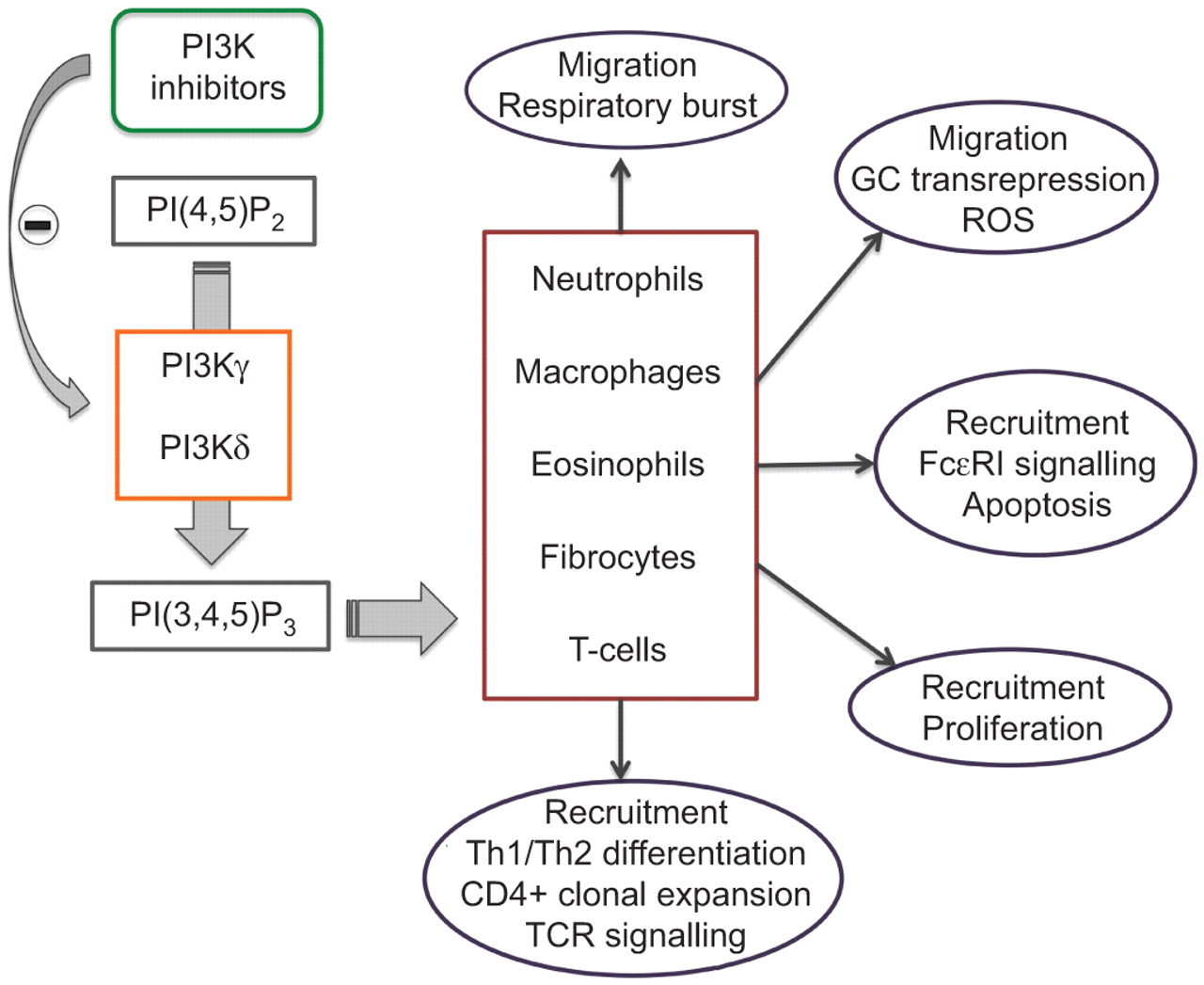
PI3K催化作用的生产phosphatidylinositol-3 4 5-triphosphate(π(3、4、5)P3),一个重要的信号转导的第二信使,并启动几个胞质事件导致细胞生长,进入细胞周期,细胞迁移和细胞存活率(84年]。这些事件的几个促炎。PI3K激活的巨噬细胞和中性粒细胞(很重要85年在慢性阻塞性肺病(),它的功能可能会改变86年]。因此,抑制PI3K被建议作为一种新颖的慢性阻塞性肺病的治疗策略。
PI3K家庭分为三个亚型(类I, II和III),根据其结构、磷脂基质和监管模式87年]。类我酶扮演了一个重要的角色在许多炎症反应的步骤(图5)。类我pi3k可以进一步催化亚基成类相关联的子类型取决于他们的IA和类IB。在哺乳动物中,类是包括三个不同的亚型:PI3Kα,PI3KβPI3Kδ。类IB是由同种型、PI3Kγ徒G protein-coupled受体下游,趋化因子受体等。PI3Kγ和PI3Kδ表达主要(但不仅限于)白细胞,导致猜测他们的主要亚型参与PI3K-mediated信号先天和适应性免疫反应,尽管最近有人建议PI3Kβ起着至关重要的作用在中性粒细胞激活免疫复合物(88年]。
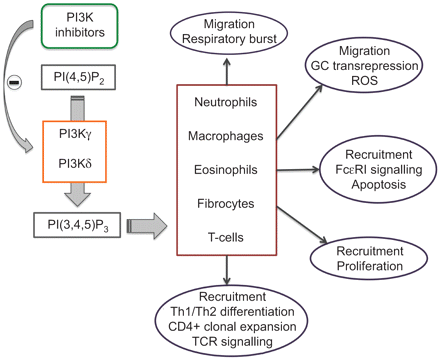
磷酸肌醇的作用的示意图表示3-kinase (PI3K)δ和PI3Kγ信号选择细胞重要的呼吸道疾病。识别:t细胞受体;ROS:活性氧;GC:糖皮质激素。
某些小分子抑制剂的PI3Kγ和δ开发(表2)[89年]。Aerolised tg100 - 115, double-selective化合物阻止PI3Kγ和PI3Kδ,抑制肺嗜中性诱发鼻内的有限合伙人和烟雾暴露在小鼠COPD模型(90年]。AS605240 PI3Kγ-selective抑制剂,减少多形核白细胞游走在体外多形核白细胞浸润,肺在活的有机体内在LPS-induced肺损伤的小鼠模型91年]。有趣的是,介入治疗与tg100 - 115抗类固醇的形式反应成功即使在慢性阻塞性肺病,诱导小鼠暴露于香烟烟雾(90年]。
尽管如此,动物缺乏功能性PI3Kγ活动显示减少主机响应链球菌引起的肺炎(92年),发现问题的重要性在慢性阻塞性肺病PI3K抑制剂。
LTB4抑制剂
LTB4导致中性粒细胞趋化作用[93年]。此外,它中性粒细胞凋亡的诱导缺陷93年]。因此,LTB拮抗作用4受体也被视为一个潜在的治疗慢性阻塞性肺病(表2)。
LTB的影响4由两个G-protein-coupled受体称为BLT1和BLT294年]。经典,BLT1 LTB提供中介4诱发炎症的中性粒细胞和巨噬细胞迁移到网站通过趋化性和粘附分子upregulation (如。Mac-1)。临床上,LTB4的主要中性粒细胞化学引诱物已被证明是呼出的气息凝结和慢性阻塞性肺病患者痰液,促进中性粒细胞的生存通过BLT1受体激活(95年]。BLT2也表达了对中性粒细胞和巨噬细胞和15-hydroxyeicosatetraenoic酸,BLT2的配体,已被证明是提高患者的慢性支气管炎和调制的LTB扮演一个角色4水平和嗜中性粒细胞趋化作用[96年]。除了BLT1和BLT2 LTB4结合并激活PPARα。这种受体的激活导致一些抗炎作用,因此有人建议,对LTB的反应4可以代表一个集成的促炎和抗炎作用,分别由细胞表面和核受体(97年]。
几个BLT1拮抗剂已经为中性粒细胞炎症的治疗开发(3,94年]。然而,到目前为止在慢性阻塞性肺病的临床研究负(98年),一般来说,他们没有被发布。有趣的是,双BLT1和BLT2受体对抗没有影响LPS-evoked嗜中性在豚鼠和香烟smoke-evoked嗜中性在小鼠和大鼠99年]。
另一个潜在的方法是防止LTB的合成4。5-lipoxygenase (LO)抑制剂,5-LO激活蛋白(瓣)抑制剂,和双5-LO /环氧酶抑制剂类抑制中性粒细胞涌入和组织水肿的口服药物动物时,可能因为减少组织LTB4水平和LTB4合成(One hundred.]。审判的八个COPD患者,5-LO抑制剂与显著提高运动能力的6分钟步行试验与安慰剂相比,慢性阻塞性肺病以及生活质量问卷分数(101年]。一项小型研究表明,皮瓣拮抗剂,BAYx1005,可以产生适度的减少一些中性的措施在COPD患者支气管炎症,但诸如FEV肺活量的端点1没有报道(102年]。然而,在最近的一次活动花絮概念验证阶段的研究中,mk - 0633年5-LO抑制剂,没有明显比安慰剂更有效改善pre-dose, pre-bronchodilator FEV1从基线在COPD患者(12周的治疗后103年]。不良反应的发生率,在任何情况下,限制5-LO抑制剂的发展(One hundred.),尽管最近记录,与慢性阻塞性肺病急性加重,需要住院期间是安全的,减少尿LTE4水平,但没有证据表明这种干预缩短住院时间104年]。
PPAR受体激动剂
PPARs ligand-activated核激素受体属于类固醇受体超家族。最初确定的作用,脂质和调节葡萄糖PPARs最近一直与其他现象的规定,包括炎症(105年]。特别是PPARs已被证明参与NF-κB转录活动的调制(106年]。PPARs transrepressive的影响是间接的,因为它们涉及干扰转录因子复合物调节炎症基因项目(106年]。
三个公认的亚型,PPARα(也称为NR1C3), PPARγ(NR1C1)和PPARδ(β或NR1C2),广泛表达(105年]。有证据表明,激活PPARγ和PPARα可能具有抗炎和免疫调节作用105年]。如前所述,LTB4直接激活PPARα[97年),但它也被记录下来,激活PPARαLTB分泌降低4(106年]。这一发现提出了一个重要的体内平衡机制的一个至关重要的促炎介质最终限制了自己的活动,从而促进解决炎症过程(107年]。
PPARγ受体激动剂,如troglitazone和罗格列酮,thiazolidinediones,抑制炎性细胞因子的释放从单核细胞和诱导细胞凋亡的淋巴细胞),这表明他们可能有抗炎作用在慢性阻塞性肺病108年]。COPD-like气道炎症动物模型的PPARγ兴奋剂,罗格列酮,抑制LPS-induced嗜中性,减少化学引诱物和生存因素(105年]。然而,在体外研究已经证明,只有大剂量的thiazolidinediones产生抗炎作用,导致问题的相关性PPARγ刺激在慢性阻塞性肺病105年]。在任何情况下,这些大剂量可能使thiazolidinediones non-PPARγ-related机制采取行动。PPARγ受体激动剂也抑制肺纤维化,因此有可能防止小气道纤维化的进展在慢性阻塞性肺病109年]。不幸的是,问题可能会妨碍这种策略已被罗格列酮治疗协会的建议增加心血管事件的风险接受2型糖尿病治疗的患者(110年]。
为了减少thiazolidinediones的潜在心血管风险,non-thiazolidinedione PPARγ配体在临床前评估。GW1929显示抗炎在保利(我:C)全身的气液界面人类支气管上皮细胞引起的类似troglitazone [111年]。
最后,考虑到减少PPARα表达式与恶病质和系统性炎症,建议PPARα受体激动剂,如安妥明和非诺贝特,可能治疗潜在的治疗慢性阻塞性肺病的系统性特征(112年]。
大环内酯类
大环内酯类引起抗炎作用可能独立于抗生素的影响(113年]。他们调节炎症和免疫反应而不影响稳态免疫(114年,115年]。
设置的慢性炎症,低剂量的大环内酯类减少促炎细胞因子和趋化因子的产生由上皮细胞和免疫细胞(113年- - - - - -115年]。这些活动在本质上是抑制和扩展粘附分子的调制必不可少的过程现场招募中性粒细胞炎症(116年]。有强有力的证据表明这些影响干扰信号通路介导的激活蛋白1,ERK1/2和NF-κB116年]。
大环内酯类拥有的能力调节上皮细胞的促炎介质的释放反应病毒(117年),导致的趋化反应显著降低中性粒细胞趋化因子(118年]。动物模型提供了进一步的证据,大环内酯类可能会阻止气道的促炎的有限合伙人的行为(119年)和改善小鼠肺气肿引起的烟的方式独立于抗生素的影响(120年]。
大环内酯物治疗诱发气道嗜中性减少在各种条件下(121年]。在慢性阻塞性肺病,抑制炎性介质的整体形象生产由慢性阻塞性肺病痰细胞似乎是定性、定量更广泛的比糖皮质激素,p38 MAPK抑制剂和PDE4抑制剂(122年]。
一些研究表明影响长期发作的频率或大环内酯物治疗普通感冒患者的慢性阻塞性肺病(123年,124年),尽管这种能力似乎是添加剂与其他传统疗法,如吸入糖皮质激素有或没有长效β2受体激动剂或长效毒蕈碱的拮抗剂。是否这是因为抗菌活性,免疫调节,或者两者兼有,仍有待确定。
大环内酯物衍生品与抗炎活动但缺乏抗菌活性会是非常有用的,因为他们将帮助避免促进耐药性。几个nonantibiotic大环内酯类,也可以称之为immunolides,现在发展为抗炎疗法(表2)[125年]。
他汀类药物
他汀类药物是3-hydroxy 3-methylglutaryl辅酶A还原酶抑制剂(β),这个属性目前广泛用于降低胆固醇的代理。最近,他汀类药物已经受到了越来越多的兴趣也为其抗炎效应(126年]。他们有能力减少生产多种促炎细胞因子(127年),还干扰白细胞粘附内皮(128年),效果会阻止白细胞进入肺实质。有趣的是,他汀类药物降低矩阵metalloproteases的生产,特别是金属蛋白酶- 1、MMP-2和MMP-9 [129年]。他们还减少氧化剂代(129年]。他汀类药物已被证明修改气道炎症动物模型和矩阵改造,特别是抑制肺气肿形成(129年]。
这些影响可以解释为什么不受控制的观测研究表明,他汀类药物降低吸烟者的肺功能下降和前吸烟者和紧急治疗和住院治疗的风险降低COPD患者,减少发病率和死亡率在慢性阻塞性肺病(130年,131年]。因此,他汀类药物在慢性阻塞性肺病管理成为有效的治疗方法。所有发表的报告支持这些代理可能有益作用在治疗慢性阻塞性肺病,但他们需要被随机对照试验证实,如国家心脏,肺,血液Institute-sponsored持续的慢性阻塞性肺病的临床试验的研究网络,STATCOPE,仔细权衡收益和风险的每日一剂量的他汀类药物在3年的一项研究中超过1000名慢性阻塞性肺病患者。尽管如此,最近的一项前瞻性研究表明,他汀类药物的使用在病人住院的COPD恶化的风险降低随后的慢性阻塞性肺病恶化和严重恶化的慢性阻塞性肺病(132年]。慢性阻塞性肺病患者他汀类药物是否有有益的影响主要是降低心血管并发症或因为他们展示一个动作直接针对肺部炎症,然而,一个争论的问题(131年]。
间接抗炎药物可能的行动
一些药物用于治疗慢性阻塞性肺病的自己并没有抗炎剂,但可以引出一个重要的间接抗炎作用(表3)。
逆转糖皮质激素抵抗
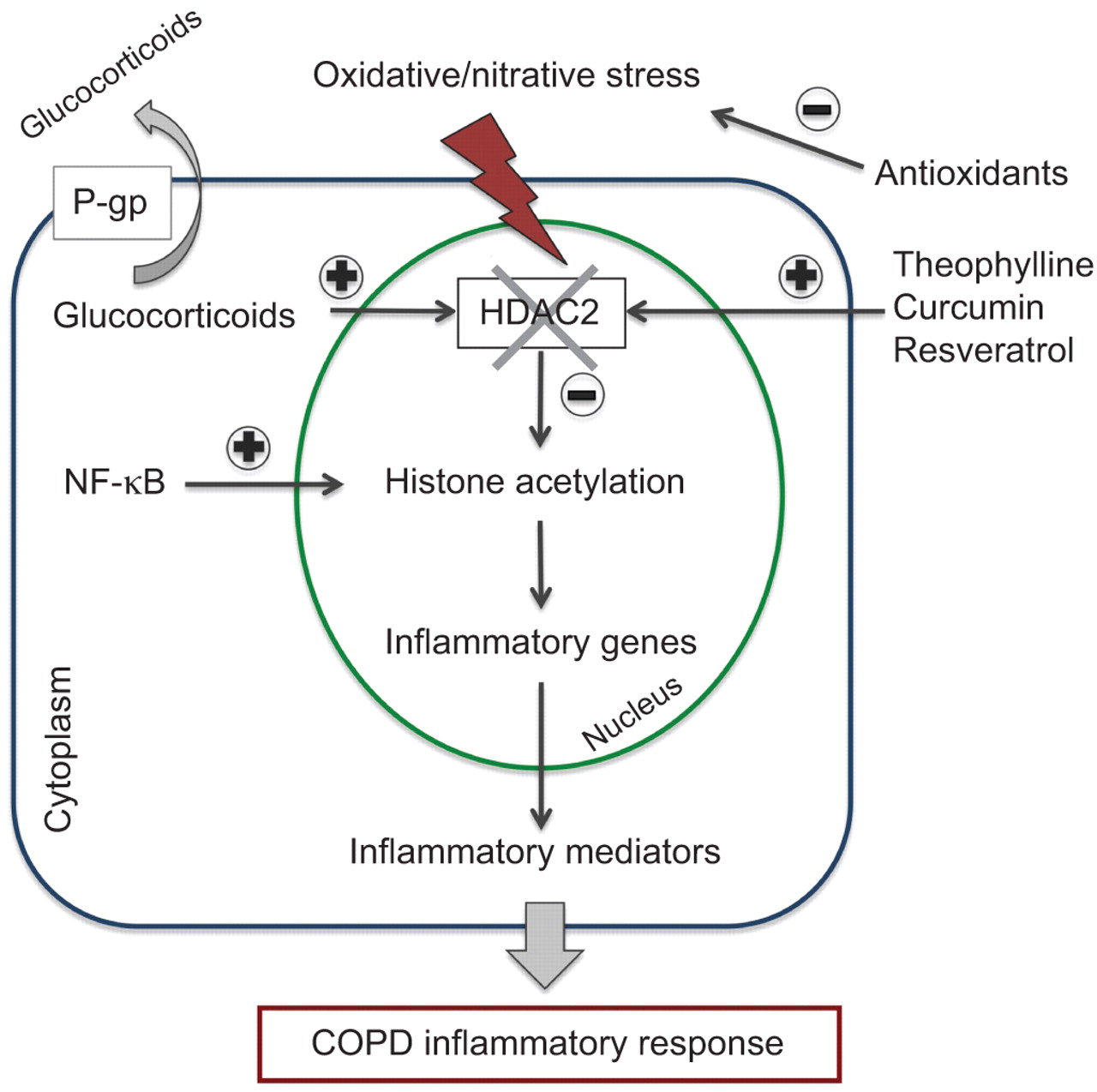
正如前面提到的,现在已经被接受,炎症在COPD患者至少部分glucocorticoid-resistant,很大程度上是因为吸烟和氧化应激损害HDAC2功能(2]。糖皮质激素抑制多种炎症慢性炎性疾病的基因被激活,激活炎症基因的组蛋白乙酰化作用通过结合配体糖皮质激素受体共激活剂分子和招聘HDAC2激活转录的复杂。在慢性阻塞性肺病,HDAC2活动和表达明显降低由于氧化/ nitrative压力使炎症成为抗糖皮质激素的抗炎作用[133年]。
在糖皮质激素抵抗的患者,药物可能逆转糖皮质激素抵抗的分子机制是被调查(图6)[133年]。可以实现选择性激活HDAC2茶碱,恢复HDAC2活动在慢性阻塞性肺病巨噬细胞恢复正常,逆转糖皮质激素抵抗。茶碱的作用的分子机制恢复HDAC2似乎通过选择性抑制PI3Kδ,在慢性阻塞性肺病患者氧化应激激活(133年]。这表明选择性PI3Kδ抑制剂也可能是有效的。由于氧化应激在减少HDAC2似乎是一个重要的机制,导致糖皮质激素抵抗,抗氧化剂还应该有效133年]。类似于茶碱,天然多酚类物质,如姜黄素和膳食抗氧化剂白藜芦醇,调解恢复糖皮质激素功能通过保护HDAC2表达和活动(134年]。
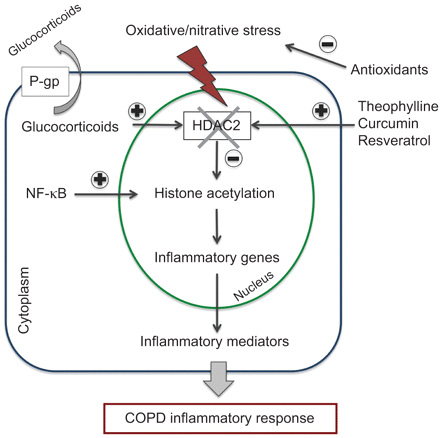
在慢性阻塞性肺疾病(COPD)患者中,慢性氧化应激激活巨噬细胞和损害HDAC2活动。HDAC2损失函数会导致增强炎症基因表达和糖皮质激素抵抗。此外,有越来越多的证据表明,膜转运蛋白的活性等22 (P-gp)可能影响细胞内的糖皮质激素浓度。P-gp挤压糖皮质激素的细胞,从而降低其细胞内的浓度。一些药物可能会逆转糖皮质激素抵抗的分子机制通过恢复HDAC2活动或抑制P-gp。NF-κB:核factor-κB。
有越来越多的证据表明,膜转运蛋白的活性,如22 (P-gp),一个跨膜蛋白从细胞挤压糖皮质激素,可能影响细胞内的糖皮质激素浓度(135年]。有人建议,高水平的表达凋亡多药耐药性基因,编码P-gp可能涉及在炎性疾病(糖皮质激素抵抗机制133年]。有几种治疗策略抑制P-gp防止糖皮质激素的流出,其中一些是基于观察维拉帕米和奎尼丁流出阻滞剂;为此一些新颖的药物正在开发(136年]。
此外,增加巨噬细胞移动抑制因子(MIF)、促炎细胞因子,有效anti-glucocorticoid效果,与糖皮质激素抵抗,所以抑制MIF的策略,包括小分子抑制剂和单克隆抗体,目前正在探索(137年]。
支气管扩张剂
加载呼吸启动炎症反应组成的高程的血浆细胞因子产生隔膜内由于增加肌肉活动和招聘和活化的淋巴细胞亚群138年]。这些细胞因子调节横膈膜肌肉拉伤也妥协横隔膜收缩,导致肌肉恶病质的发展。此外,他们可能有系统性影响,动员从肝脏葡萄糖和游离脂肪酸从脂肪组织极力呼吸肌肉工作(138年]。有人建议,支气管扩张药有可能减少炎症影响电阻减少动态的恶性通货膨胀和呼吸(138年]。
不幸的是,研究β的影响2受体激动剂的收缩性隔膜产生争议的结果(139年,140年]。此外,尽管tiotropium,长效antimuscarinic代理,可以维持肺恶性通货膨胀显著减少,因此,减少加载呼吸(141年),临床试验与这个代理还没有令人信服地证明了抗炎效应(142年]。同时,研究的肌肉收缩能影响methylxanthines提供一些矛盾。氨茶碱增加横隔膜收缩性,扭转膜片疲劳(143年此外,)和小剂量茶碱在慢性阻塞性肺病(抗炎作用144年]。然而,这些影响没有观察到所有的调查人员,也有怀疑,他们将减少加载呼吸(144年]。这些发现问题的重要性在减少炎症引起的支气管扩张剂加载呼吸。
抗氧化剂的策略
香烟和烟草烟雾/生物质fuel-induced氧化和醛/羰基应激在慢性阻塞性肺病与炎症密切相关。因此,代理可以抑制活性氧的生成或可以抵消这些物种或有可能间接影响炎症(145年]。
各种抗氧化剂之间尝试过到目前为止,硫醇抗氧化剂和黏液溶解的药物,和膳食多酚已报告增加细胞内硫醇状态以及感应谷胱甘肽的生物合成。这样的海拔在硫醇状态进而导致自由基解毒和氧化剂以及抑制持续的炎症反应。谷胱甘肽的生物合成的诱导物(Nrf2活化剂)、抗氧化剂维生素、旋转陷阱,超氧化物歧化酶、谷胱甘肽过氧化物酶的模拟和脂质过氧化作用和蛋白质羰基化抑制剂/阻滞剂也被证明有有利影响通过抑制香烟烟雾诱发炎症反应和其他羰基/氧化应激细胞的改变(表3)[145年]。
由于各种氧化剂,自由基和醛与COPD的发病机制,结合各种抗氧化剂的影响随着硫醇,旋转陷阱,脂质过氧化反应/蛋白质羰基化抑制剂/阻滞剂,或酶模拟是一个有趣的命题在COPD患者值得研究。抗氧化剂(如。硫醇和其他分子)结合抗炎药/ PDE4抑制剂/ Sirtuin1活化剂,支气管扩张剂,类固醇,抗生素,和他汀类药物145年]。
蛋白酶抑制剂
中性粒细胞、巨噬细胞和细胞毒性t淋巴球释放蛋白水解酶的数组,包括丝氨酸(弹性蛋白酶,蛋白酶3)、半胱氨酸(组织蛋白酶S)和-金属蛋白酶(MMP-12)。这些蛋白酶分裂细胞外基质成分,弹性纤维和胶原蛋白,产生弹性蛋白片段或collagen-derived肽如proline-glycine-proline,已被证明是对单核细胞趋化,前驱细胞巨噬细胞和中性粒细胞(146年]。
蛋白水解级联的台阶存在多个潜在的治疗目标(147年]。开发了新的化合物与每一个步骤(表3)。胶原蛋白和弹性蛋白处理每个特性基质金属蛋白酶活性蛋白酶。非特异性MMP抑制剂如ilomastat (gm - 6001), marimastat、rs - 113456或因此cp - 471474可以抑制促炎信号由胶原蛋白和弹性蛋白降解(表3)。此外,特定的拮抗剂的胶原蛋白和弹性蛋白分解显示承诺在临床前工作。
许多合成中性粒细胞弹性蛋白酶(NE)抑制剂开发潜在的治疗药物(表3)[148年]。这些包括不可逆抑制剂和可逆的抑制剂。低分子量的问题之一可逆抑制剂是它们能释放NE,允许它破坏组织。虽然不可逆抑制剂已被证明有效的功能在活的有机体内在仓鼠减少许多气管内的管理NE的影响,其毒性防止临床使用。Sivelestat,在日本和韩国市场销售治疗急性肺损伤与全身炎症反应综合征有关,和ono - 6818开发并看到了好坏参半的结果在人类研究中,而其他NE抑制剂在临床前或第一阶段试验停止由于各种原因(149年]。有趣的是,3个月的治疗AZD9668没有改善肺功能、呼吸系统症状和体征或生活质量得分当添加到布地奈德/ formoterol维持治疗在慢性阻塞性肺病患者150年]。NE抑制剂在慢性阻塞性肺病的缺乏影响可能是由于中性粒细胞的作用比此前认为的COPD的发病机制不太重要(150年]。另外,添加NE抑制剂可能导致没有进一步的好处除了已经获得了与常规维持治疗。
结论
在2004年,美国胸科学会/欧洲呼吸学会指南(188bet官网地址151年)发现了一个迫切需要开发代理商,抑制炎症与慢性阻塞性肺病和防止疾病进展。在过去的几年,一些新的潜在目标已确定和小说代理为这些新目标,以及为已知的目标了。这些干预措施已经有效的动物模型,但翻译没有直接对人类和动物之间有重大差异的数据和人体试验(152年]。
有限的一个可能的解释明显功效的抗炎治疗在慢性阻塞性肺病慢性阻塞性肺病已成为临床上明显的时候,该疾病已经比较先进,与不可逆转的实质损害,从而限制抗炎治疗的好处,尤其对肺功能(113年]。因此,差异的原因在动物上的数据和人体试验可能涉及,在很大程度上,无法在实验室动物产生严重的慢性阻塞性肺病。此外,充分发展新型抗炎剂可能会有问题,因为目前使用的临床评估COPD患者并不代表的炎症过程153年]。
由于这些原因,这并不奇怪,只有药物之一,roflumilast,口服PDE4抑制剂,已达到市场治疗慢性阻塞性肺病的,而其他所有的人类新的药物抗炎方法测试已经发现无效或背负重大的副作用。很明显,尽管在这一领域所取得的进展,仍有显著的差异在我们的理解。我们所真正了解的是,小说治疗慢性阻塞性肺病的发展,除了支气管扩张剂,仍然是一个挑战。
脚注
感兴趣的语句
没有宣布。
- 收到了2011年12月6日。
- 接受2012年3月5日。
- ©2012人队
引用
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}