





文摘
呼吸衰竭发生主要是由于肺衰竭导致低氧血或泵失败导致肺泡肺换气不足和血碳酸过多症。Hypercapnic呼吸衰竭机械缺陷,可能是由于中枢神经系统抑郁,能源需求和供给的不平衡和/或改编的中央控制器。
Hypercapnic可能发生呼吸衰竭严重,在不知不觉中或严重慢性二氧化碳潴留。在这些条件下,病理生理,公分母是减少肺泡通气对于一个给定的二氧化碳生产。
急性hypercapnic呼吸衰竭通常是由缺陷引起的中枢神经系统,神经肌肉传递障碍、机械缺陷的胸腔和呼吸肌肉的疲劳。
负责慢性的病理生理机制二氧化碳潴留尚未明确。最具吸引力的假设这个障碍是“自然的智慧”的理论。患者面临负载有两个选择,要么努力推动为了维持正常动脉二氧化碳和氧气紧张的代价最终成为疲劳和疲惫或呼吸每分通气量较低,避免呼吸困难,疲劳和疲惫但肺泡通气量的减少。基于最近的作品,喜欢的假设是一个阈值吸气负载可能存在,超过时,会导致肌肉损伤,因此,一个适应性反应引起预防和/或减少这种损害。这包括细胞因子的生产,反过来,调节呼吸控制器,直接通过血液或可能是小传入或通过-肾上腺轴。调制模式的呼吸,然而,最终导致肺泡肺换气不足和二氧化碳潴留。
本研究在胸腔基金会的支持下,雅典,希腊。
呼吸衰竭是一种状态,呼吸系统失败的一个或两个气体交换功能,即。氧化和/或消除二氧化碳的混合静脉血。这是通常定义为一个动脉氧张力(P啊,一个2)< 8.0 kPa(60毫米汞柱),一个动脉二氧化碳张力(P,有限公司2> 6.0 kPa)(45毫米汞柱)或两者兼而有之。因此,呼吸衰竭的诊断实验室,但强调的重要的一点是,这些截止值不是刚性;他们只是作为一般指南结合历史和病人的临床评估。
呼吸系统可以表示由两部分构成:肺,即。gas-exchanging器官,通风肺部的泵1。泵由胸壁,包括呼吸道肌肉、呼吸控制器在中枢神经系统(CNS)和通路连接中央控制器与呼吸的肌肉(脊髓和周围神经)。的每一部分系统的失败会导致截然不同的实体(图。1⇓)。一般来说,失败引起的肺部各种肺部疾病(如。肺炎、肺气肿和肺间质疾病)导致低氧血与血碳酸正常或低碳酸血(hypoxaemic或I型呼吸衰竭)。泵的故障(如。药物过量)导致肺泡肺换气不足和血碳酸过多症(hypercapnic或II型呼吸衰竭)。虽然有同时代的低氧血,通气失败是增加的标志P,有限公司2。毫无疑问,这两种类型的呼吸衰竭可能共处在同一个病人,,例如,慢性阻塞性肺病(COPD)患者和二氧化碳潴留,或在那些严重的肺部水肿或哮喘危机,谁先开发低氧血,随着疾病的持续或发展,出现血碳酸过多症。
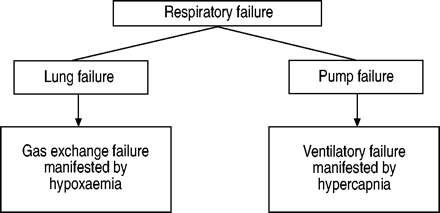
类型的呼吸衰竭。呼吸系统可以被认为是包括两个部分:1)肺;2)泵。
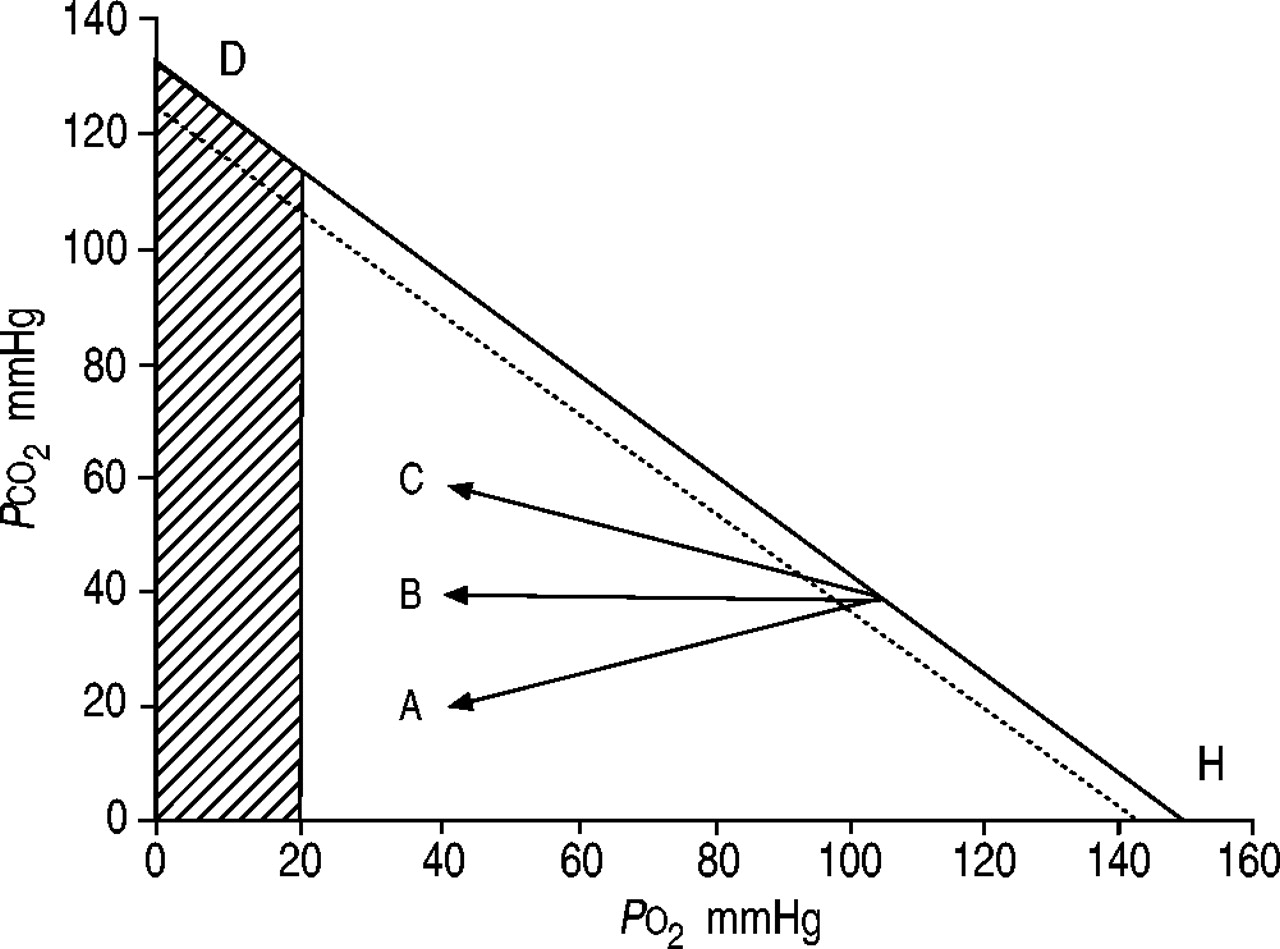
并给出了各种类型的呼吸衰竭气体中的紧张关系图(图2所示⇓),这说明了各种途径。实线代表一个呼吸交换比率为0.8。并行虚线显示相应的P啊,一个2和P,有限公司2与肺泡动脉氧差异(D一个必经aO20.67∼kPa)(5毫米汞柱),发生在正常的肺。当一个正常换气过度,肺泡氧气(P啊,一个2)和二氧化碳(P,有限公司2)紧张和P啊,一个2和P,有限公司2向下移动方向的斜率表示字母H,上升P啊,一个2和P啊,一个2和瀑布P,有限公司2和P,有限公司2。当发生肺换气不足时,在正常的话题,例如,由于药物过量P啊,一个2和P啊,一个2和P,有限公司2和P,有限公司2如图所示方向上升斜率的字母D,下跌P啊,一个2和P啊,一个2和上升P,有限公司2和P,有限公司2。可以看出,在正常的肺血碳酸过多症发生时(如肺泡肺换气不足由于中枢神经抑郁)的情况下,P啊,一个2不能降至非常低的水平。例如,当P,有限公司2增加从(40毫米汞柱)的5.3∼10.6 kPa(80毫米汞柱),P啊,一个2减少从13.3∼(100毫米汞柱)∼8.0 kPa(60毫米汞柱)。假设D一个必经aO20.67 - -1.3 kPa(5 - 10毫米汞柱)P啊,一个25.3∼-6.7 kPa(40 - 50毫米汞柱)。因此,当肺是正常的,严重程度的肺泡肺换气不足,导致二氧化碳潴留,不是与过度的低氧血有关。在肺部疾病,然而,由于增加了D一个必经aO2导致动脉低氧血,同样的条件。箭头Α,如图2所示⇓,显示了一个大D一个必经aO2(箭头Α之间的水平距离和肺泡D量H行),这是一般观察患者的肺炎、肺不张或急性呼吸窘迫综合征(ARDS)。在这些患者中换气过度会导致非常低P,有限公司2。行Β描绘了通路的间质性肺病患者或纯肺气肿。直线C描绘了一个混合态的肺病患者(低氧血)和肺泡通气不足(V”一个)。在严重的情况下(箭头的提示),低氧血是占主导地位,尽管血碳酸过多症,情况肯定比在纯肺换气不足平等更危险P,有限公司2。患者通常达到C从线Α或B . COPD患者终末期肺间质疾病一直是沿着箭头B很长一段时间箭头移动到C由于肺泡肺换气不足。同样,气体交换异常患者,如箭头所示Α(急性哮喘发作或肺部水肿),可能转向箭头B或C作为中央控制器或呼吸道肌肉,或两者兼而有之,成为无法保持足够的通风。
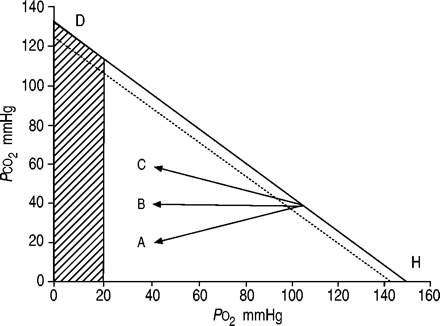
肺泡氧气之间的关系(PO2)和二氧化碳(P有限公司2)紧张当呼吸空气(- - - - - -;吸入氧气比例21%)。直线的斜率取决于呼吸交换比率;在本例中,它是假定为0.8。动脉PO2(═)小于肺泡PO2;认为这是5毫米汞柱的区别。量C:方向肺泡气体紧张局势的变化各种肺部疾病。┘:动脉PO2通常与长期生存不兼容(1毫米汞柱= 0.133 kPa)。
呼吸衰竭的病理生理学
Hypoxaemic (I型)呼吸衰竭
四个病理生理机制占低氧血出现在各种各样的疾病:1)通气/灌注不平等,2)分流增加,3)扩散障碍,和4)肺泡肺换气不足2。通气/灌注失配是最常见的机制和发展时减少通风正常灌注区域或者当有肺部区域更大比灌注减少通风。分流,肺内的或心脏内的缺氧混合静脉血绕过通风肺泡,导致“静脉剂”。疾病,增加氧气的扩散途径从肺泡空间到肺毛细血管,降低毛细血管表面积或缩短运输时间的血液通过肺部毛细血管阻止完整的肺泡与肺毛细血管血液氧平衡。
没有潜在的肺病,低氧血伴随肺换气不足的特点是正常的D一个必经aO2。相比之下,障碍,其他三个机制是手术的特点是拓宽肺泡/动脉梯度导致严重的低氧血症。
虽然变化V”一个可以改变P,有限公司2大大,事实并非如此P啊,一个2。增加V”一个适度增加P啊,一个2。由于s形oxyhaemoglobin离解曲线的形状,有增加通风对血氧饱和度的影响是最小的P啊,一个27.3 - -8.0 kPa(则高达55 -毫米汞柱)。低氧血造成通气/灌注不平等或扩散异常可以很容易地纠正补充氧气的启发,尽管非常高浓度的氧气不能启发正确增加引起的低氧血纯粹的分流。
Hypercapnic (II型)呼吸衰竭
呼吸系统方程
二氧化碳的体积消除每分钟(在一个稳定的状态等于身体产生的(V”有限公司2)依赖于肺泡气体和οn中的二氧化碳浓度V”一个。这是显而易见的,因为开展航空不交换气体。因此V”有限公司2=V”一个×肺泡有限公司2浓度或肺泡有限公司2浓度=V”有限公司2/V”一个。肺泡有限公司2浓度CO的浓度2在肺泡气体。气体浓度转化为气体压力(P气)的方程:P气体(毫米汞柱)=(%浓度×(气压加压水蒸汽))/ 100。在海平面上,气压是670和水蒸气压力在37°C是47毫米汞柱。由此可见,P气体浓度(毫米汞柱)= %×713/100。
通过使用因子k(0.863),比例常数,获得了“呼吸方程”,有关V”一个来P,有限公司2: 自V”一个=V”E−V”ds,在那里V”E每分通气量,V”ds死亡空间通风,这种关系可以表示为:
自V”一个=V”E−V”ds,在那里V”E每分通气量,V”ds死亡空间通风,这种关系可以表示为: 在哪里VT是潮汐卷和fR呼吸频率。
在哪里VT是潮汐卷和fR呼吸频率。
方程2的州P,有限公司2如果上升V”有限公司2增加(如。高热)恒定V”一个,或者当一个常数V”有限公司2,V”一个减少由于:1)上升V”ds/VT(通过增加V”ds,降低VT或者两者都有),2)下降V”E和3)的增加V”ds/VT在减少V”E3,4。
在日常临床实践中,随着病人变得hypercapnic,通常超过一个因素导致的P,有限公司2。
二氧化碳生产
对于一个年轻的成年人,V”有限公司2∼200毫升·分钟吗−1(或110毫升·m2在男性和96毫升·m2在雌性)。V”有限公司2增加在高热∼14%每升高一摄氏度温度上升,特别是在肌肉活动。在吸气电阻呼吸,呼吸的肌肉,在这方面,可能显示V”有限公司2700 - 800毫升·分钟−15。以同样的方式,颤抖或肌张力增加,如破伤风发生,导致过度V”有限公司2增加到三倍,而肌肉锻炼可能会增加V”有限公司2> 10倍。在正常情况下,增加V”有限公司2是由中枢神经系统早期发现的,然后很容易弥补增加V”E保持一个正常的P,有限公司2。然而,如果一个病人的通气能力受损,增加V”有限公司2极大地强调了通气系统,导致增加P,有限公司2。
肺泡通气
方程1和2暗示,在恒定V”有限公司2和一个给定的V”ds,V”一个变化时VT或fR不同的恒定或减少总通风。这意味着有四种可能:1)总通风与降低不变fR总通风与增加,2)不变fR3)降低总通风与降低fR或4)下降VT。
总通风和不变的条件下降低了fR,因为V”E保持不变,VT必须增加。这将会减少V”ds/VT,从而增加V”一个和减少P,有限公司2。
总通风和不变的条件下增加fR,因为V”E保持不变,VT必须减少。然而,这样的改变,增加了V”ds/VT率,因此,V”一个减少和P,有限公司2增加。在临床设置,快速浅呼吸很可能解释在COPD患者二氧化碳潴留。
的条件下减少总通风和减少fR,减少fR孤独,而不影响V”ds/VT,导致了一定的下降V”一个由于减少V”E。
的条件下减少总通风和减少VT,有减少V”E减少造成的VT(没有减少fR),这导致增加V”ds/VT,因此,在上升P,有限公司2。因此,下降V”一个预计将比在上述情况下更为明显。
病理生理学的通气泵衰竭
有三个泵衰竭导致血碳酸过多症的主要原因6。1)呼吸系统的输出中心控制的肌肉可能不足(麻醉、药物过量、髓质)的疾病,导致中央呼吸驱动的需求不足,或呼吸中心可能本能地修改它们的输出以防止呼吸道肌肉拉伤和避免或推迟疲劳。2)有机械在胸壁缺损,在连枷胸,一样的疾病(格林-巴利综合征)和前角神经细胞(小儿麻痹症),或呼吸道疾病的肌肉(肌肉疾病)。严重的恶性通货膨胀,与平隔膜和减少机械作用的吸气肌肉,如急性哮喘发作,是一种最常见的原因受损的吸气肌肉的力学性能。3)吸入过多负荷下工作时,吸气肌肉会变得疲劳,即。他们变得无法继续产生足够的胸膜压力尽管适当的中枢呼吸驱动和一个完整的胸壁。
很明显,当激活中枢神经系统的不足,要么暂时(如。麻醉和过量)或永久(如。疾病的髓质),呼吸的努力是不够的,肺换气不足就会随之发生。
电动机输出来自需要转移到呼吸中枢神经系统肌肉,这一过程需要解剖和功能脊髓的充分性,周围神经和神经肌肉接点。任何障碍这个途径不足导致通货膨胀的胸腔产生负压不足,这是必不可少的空气流进入肺部。胸壁的机械缺陷(连枷胸,脊柱后侧凸和恶性通货膨胀)是实体,使肺泡肺换气不足,因为他们在吸气肌肉施加额外的工作,取代了不合规的胸壁和肺。
恶性通货膨胀以来,常见疾病的特点是气道阻塞和损失的弹性反冲的肺,有多个负面影响吸气肌肉功能,值得单独讨论。
自然对人类呼吸,吸气肌肉必须产生足够的力量来克服弹性和呼吸系统的电阻负载。此外,吸气肌肉应该能够维持上述负载随时间变化和调整V”E在这样一个有足够的气体交换。疲劳是呼吸肌肉无力继续产生足够的压力来维持V”一个6。疲劳应该区分开来的弱点,这是一个固定的减少产生不可逆的武力休息,虽然肌肉无力可使肌肉疲劳。
疲劳时发生呼吸肌肉的能源供应不符合要求。因素诱发呼吸道肌肉疲劳是那些吸气肌肉增长的能源需求和/或减少能源供应7。能源需求是由呼吸和吸气肌肉的力量和效率(图3所示⇓)
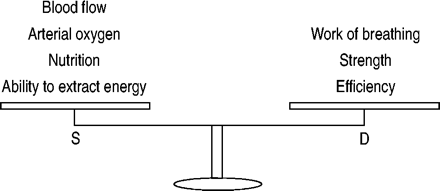
呼吸肌肉的耐力是由能源供应之间的平衡(S)和要求(D)。通常情况下,供应满足需求和大量储备的存在。当这种平衡重量的要求,最终呼吸肌肉变得疲劳,导致无法维持自主呼吸。
呼吸的工作增加比例与平均压力由每个呼吸的吸气肌肉(意思是潮汐压力(P我)),表示为最大吸气压力的一小部分(P我,马克斯),V”E工作周期(吸气时间(t我)/总呼吸周期(t合计))和平均吸气流量(VT/t我)6。
P我如果增加弹性(僵硬的肺,肺水肿)或电阻(气道阻塞、哮喘)负载对吸气肌肉增加。·鲁索斯et al。8直接相关的P我/P我,马克斯隔膜的时间可以维持负载对它(持续时间)。的临界值P我/P我,马克斯可以无限期地在生成功能余气量(FRC) 0.60∼。更大的P我/P我,马克斯耐力时间呈负相关。的临界值P我/P我,马克斯增加当呼气肺体积增加。实际上,当肺容积增加从FRC FRC + 50%的吸气量,关键的价值P我/P我,马克斯和持续时间减少到非常低的值,25 - 30%P我,马克斯。Bellemare和Grassino9还发现,最大压力时,可以无限期持续减少t我/t合计增加和建议的产物P我/P我,马克斯和t我/t合计定义了一个有用的“紧张时间指数”,持续时间有关。每当紧张时间指数低于临界值隔膜(0.15),负载可以持续下去。
弱肌肉需要更多的能量与它的最大能源消耗执行给定的工作。力由一个足以产生骨骼肌疲劳是一个函数的最大力量,肌肉可以开发。任何条件,降低了最大力量减少肌肉的力量和容易疲劳。这些条件包括萎缩(长期机械通气的可能的结果),不成熟,神经肌肉疾病和性能在一个低效的肌肉的长度/张力特性的一部分10在急性恶性通货膨胀的状态,在此期间隔膜和肋间肌肉长度较短的工作。
最后,肌肉效率、外部工作的比例进行能源消耗,能源需求的一个重要因素。吸气肌肉效率下降患者的恶性通货膨胀。它已经表明,氧呼吸的同样的工作,成本是肺气肿患者明显高于正常人11。这一切发生的时候,在肺气肿的病人,因为有些吸气肌肉合同同分异构地(他们消耗能量,但不执行工作)或吸气肌肉操作效率低下的一部分力量/长度关系:需要一个更有力的收缩产生一个给定的压力变化,和一个更大程度的激发需要开发一个给定的力量。因此这两种情况导致能源消耗压力对于一个给定的发展12。
因素确定吸气肌肉能量可用肌肉血流量,Ca, O2和血液底物浓度以及肌肉中提取能量的能力(图3所示⇑)。
横隔膜的血液流动本质上是由灌注压力,这是心输出量和外周血管阻力的函数,和肌肉的血管阻力,收缩的强度和持续时间的函数13。作为动物模型被描述,减少心输出量伴随心原性或感染性休克是引起呼吸道疲劳导致严重的肺泡肺换气不足,呼吸徐缓和呼吸停止14,15。能源供应吸气肌肉还取决于肌肉的能力来增加血液流动与增加并行工作。隔膜有更大的能力比其它骨骼肌增加血流量16。然而,吸气肌肉血流量的数量可以增加可能会影响到肌肉收缩的强度和持续时间。如果呼吸肌肉保持简约在整个呼吸周期,在哮喘的发生17,整个血液流向肌肉可能少于所要求。此外,血红蛋白浓度和oxyhaemoglobin饱和度影响肌肉有氧能量供应,因此它的耐力。
条件的特点是肌肉无法提取和使用能源,如败血症或氰化物中毒,或减少能源存储和糖原耗竭,在极度空虚,可能导致呼吸道肌肉疲劳。
很明显从上面的讨论,疲劳可能发生在各种各样的临床实体单独或结合导致呼吸肌肉的能源供应和需求之间的不平衡。不管原因是什么,众所周知,疲劳的特点是力输出的损失18,导致无法呼吸的肌肉充分发展P我在潮汐呼吸,随之减少VT和V”E血碳酸过多症。
当装载呼吸肌肉广泛,然而,很可能反馈机制修改中央驱动,通过施加“中央智慧”,改变了通气模式和服务在减少负载,减轻疲劳,因此保护通气泵从疲惫,毫无疑问,是一个终端事件。
虽然没有数据从病人证实存在的“中央智慧”在通气失败,有足够的证据可以支持这一观点。减少的事实VT电阻后可以立即恢复正常呼吸动物纳洛酮19或双边颈迷走神经切断术20.,以及大多数hypercapnic COPD患者可以达到血碳酸正常通过自愿增加通风,意味着,尽管受试者可以增加通风,他们选择不这样做。
事实上,改变呼吸的模式可能会出现由于加载在动物身上19,正常的话题21和病人在断奶期间试验22。
急性和慢性呼吸衰竭患者以及正常受试者和动物受到疲劳呼吸负荷往往采用快速浅呼吸,减少组成VT和增加fR,而V”E保持不变或略有增加。尽管这种模式可能不是有效的气体交换,它可能减少肌肉减少负载P我发达,从而防止疲劳的发生23- - - - - -26。此外,在稳定的COPD患者和二氧化碳潴留,这种模式的呼吸可能足以使隔膜收缩低于疲劳门槛23,27。
的神经生理学机制导致呼吸模式的改变是不能很好地阐明。Chemosensitivity-induced改变呼吸活动似乎没有解释。缺氧,hypercapnia-induced减少呼气时间(tE)是不成比例大于减少t我,所以t我/t合计增加。此外,VT/t我和VT增加而不是减少28。
快速浅呼吸可能由激活迷走神经刺激受体的航空公司29日,也可能代表一个行为反应减少呼吸困难的感觉30.。
反射来自机械在胸腔肌肉萎缩和隔膜(腱器官、主轴器官和类型III和IV结局)可能发挥作用在塑造快速浅呼吸模式。在深深犀牛动物,肋间肌肉的伸展或隔膜张力的增加可能会突然终止的灵感31日。激活内源性阿片样物质途径也被假定改变呼吸的模式,或许是一种机制,通过这种机制可能减少呼吸困难的感觉19,32- - - - - -35。
Small-fibre传入纤维广泛涉及中央呼吸系统的响应输出等长期的压力冲击,缺氧、酸中毒和剧烈运动15,36,37。可能,在加载呼吸,传入,通过小纤维,调节内源性阿片类物质作为减少呼吸困难的适应性反应,避免或延缓呼吸肌肉疲劳的发生38。
无论机制,然而,这种策略的限制,快速浅呼吸V”ds/VT增加(见呼吸系统方程部分)血碳酸过多症的恶化。
虽然失败的肺通气的泵的主要是低氧血和失败导致血碳酸过多症,必须强调,有重要的这两个实体之间的交互。此外,失败的一部分(肺)通常是紧随其后的是第二次的失败(通气泵)。呼吸道疾病导致低氧血通常特点是肺力学异常,这种情况伴随着增加呼吸(电阻或弹性)的工作,因此,能源需求。考虑到这一事实的能量可以减少由于低氧血,得出肺部疾病可能导致肌肉疲劳和通气失败通过需求和供给之间的不平衡。
同样,患者的疾病包括通气泵(肌肉疾病)和与血碳酸过多症通常呈现的特点是不能咳嗽,可能导致分泌物的积累和肺不张,情况最终加重通气/灌注不平等与低血氧症的结果。
肺恶性通货膨胀
在正常受试者静止,呼气肺体积(FRC)对应于放松量(Vr呼吸系统的),即。的肺容积总呼吸系统的弹性反冲压力为零。肺恶性通货膨胀的定义是FRC在预计正常价值的增加。这可能是由于增加了Vr由于肺的弹性反冲的损失(如。肺气肿),或由于动态肺恶性通货膨胀,发生当FRC超过Vr39。
不管它的起源,恶性通货膨胀对肌肉功能诸多限制。骨骼肌的吸气肌肉,至于其他,有一个最优长度的最大力量。这个长度对应密切FRC或降低肺容积6。恶性通货膨胀,呼吸肌肉纤维缩短,因此可用的力量或压力发达对于任何给定水平的激励却降低了。这导致的情况,对于一个给定的压力所必需的足够的摆动V”一个,更大的激发,因此更大比例的最大压力,是必需的。为给定的工作负载,这就增加了能耗和肌肉效率因此减弱。
隔膜进一步受到恶性通货膨胀引起的几何畸变的吸气肌肉。隔膜是扁平的,假设一个半径为无穷大,拉普拉斯定律(P迪= 2T迪/ R迪,在那里P迪是transdiaphragmatic压力,T迪产生的切向拉力隔膜和R迪隔膜的曲率半径),隔膜不再是能够有效地把紧张的压力40。除此之外,随着隔膜趋缓,欧元区的反对会降低。该区域的区域毗邻侧胸壁的肋隔膜。在正常受试者,当隔膜收缩,增加腹压胸腔穿过该区域传播,从而促进胸扩张。因此,随着肺容积增加,横隔膜收缩胸壁扩张的贡献是最小化。
此外,恶性通货膨胀改变了膈肋的空间定位和脚纤维,迫使他们在系列和垂直地安排对胸壁。在灵感,这些面向垂直地收缩纤维内矛盾运动的结果低胸腔(胡佛的标志)。
与隔膜,胸腔的长度/张力特征和几何位置的吸气肋间的恶性通货膨胀期间或配件的肌肉处于劣势6。
的呼吸,几乎总是增加了异常肺力学导致恶性通货膨胀,进一步增加了恶性通货膨胀本身。因此,能源需求增加,而能源供应可能有限由于呼吸肌肉持续收缩。增加工作的结合,降低强度,降低效率和降低能源供应在恶性通货膨胀使呼吸肌肉更大风险,使他们特别容易疲劳。
在临床条件下通气失败
Hypercapnic可能发生呼吸衰竭严重,在不知不觉中或严重慢性二氧化碳潴留。
血碳酸过多症急性发作
病理实体导致急性二氧化碳潴留是中枢神经系统的解剖和功能缺陷,损伤的神经肌肉传输和胸腔的机械缺陷,以及条件导致呼吸肌肉的疲劳(表1所示⇓)。机制负责二氧化碳潴留都减少V”E和增加V”ds/VT。
抑郁症的中枢神经系统药物、感染或头部外伤导致肺换气不足由于呼吸道受损。
机械缺陷的胸壁(连枷胸和急性恶性通货膨胀),神经肌肉疾病(双边膈麻痹、重症肌无力、肉毒中毒和格林-巴利综合征)和箭毒等药物可能导致急性血碳酸过多症。
急性血碳酸过多症,最终疲劳可能发生在每一个临床条件的特点是呼吸的机械负荷,增加,因此,在能源需求无法满足的能源供应。在这些条件下,中枢神经系统可能会条件反射性地调整其输出信号,以避免在肌肉细胞完成至关重要的化学物质的损耗18和/或公开的疲劳。通过这种方式,VT减少递减t我为了使P我在能源需求减少,因此每呼吸(紧张时间每减少呼吸指数)。此外,小潮呼吸,呼吸肌肉运作的更优的长度并不显著影响其几何。减少VT是补偿,至少在一开始,通过增加fR这V”E维持或增加。这种情况会导致快速浅呼吸41,42,增加V”ds/VT血碳酸过多症。
这fR然而,不再是最优的,相同的V”一个,能源需求增加。因此,尽管缩短t我似乎是一个更好的选择的能源需求,如果加上高fR和可能的能源供应不足,发生在剧烈的收缩,这可能会导致肌肉疲劳。压力产生的吸气肌肉然后随随之减少VT和V”E和增加V”ds/VT42,随后的减少V”一个这是伴随着增加P,有限公司2。在稍后的阶段,t我增加一遍又一遍fR逐渐减少,导致进一步下降V”E43。在极度疲劳,每呼吸中枢神经系统降低了输出信号,导致呼吸停止14。
因此肺泡肺换气不足与顺向血碳酸过多症的结果减少紧张时间指数,这可能是由于肌肉疲劳或适应前的中枢神经系统发展的明显的疲劳。
在急性哮喘发作,严重的呼吸道梗阻、快速浅呼吸与增加弹性和电阻吸气相关联的工作负载的增加呼吸,因此能源需求。呼吸肌的力量和效率降低恶性通货膨胀,以及血液供应受损由于强烈的肌肉收缩,可能会导致降低了P我,马克斯,而与此同时,P我是增加了。增加P我/P我,马克斯导致呼吸困难30.,可能,如果一个临界值交叉,疲劳。这种情况下部队肺泡通气减少VT作为肌肉的保护机制,因为有足够的证据表明剧烈呼吸引起肌肉拉伤44,45,或者由于疲劳的肌肉。
在noncardiogenic (ARDS)和心脏发生的肺部水肿,能源需求增加是由于增加了弹性工作和换气过度,而能源供应减少是由于低氧血在前,和低氧血以及低心输出量是后者。在这种情况下,呼吸肌肉可能会失败。
急性hypercapnic呼吸衰竭也可能在急性呼吸衰竭患者发展断奶期间接受机械通气的审判。病理生理途径导致二氧化碳潴留在这组病人更好的学习,因为,在其他临床条件下,它是困难的,如果不是不道德的,延迟治疗干预措施以文档的生理参数。
在第一个相关的和非常重要的研究中,由科恩et al。43,发现大多数患者没有断奶的审判提供tachypnoea,酸中毒和呼吸肌肉的疲劳。上面的作者,以及其他随后,提出疲劳可能是最后共同通路导致停药期间hypercapnic呼吸衰竭机械通气22。关于这个问题的进一步研究表明,快速浅呼吸(随之上升V”ds/VT),以及一个显著增加呼吸肌肉的弹性和电阻负载的主要原因是二氧化碳潴留在这些病人22,24。虽然有一些争议的确切作用在脱机失败吸气肌肉疲劳,有足够的数据提供重要支持的疲劳。戈德斯通et al。46在断奶期间测量最大松弛率,观察患者的下降没有断奶,一个发现表明疲劳过程开始在书中呼吸的肌肉。此外,使用标准类似于科恩et al。43,Brochardet al。47发现患者未能被断奶展出肌疲劳的迹象在自主呼吸减少紧随其后VT,增加fR血碳酸过多症和发展。停止使用呼吸肌肉的萎缩,短期和长期机械通气后记录的条件48的灌注减少,营养不良,经常会出现在这些病人,并减少肌肉的力量和效率由于恶性通货膨胀是条件,减少呼吸道肌肉能力。加上机械负荷增加,这些情况导致呼吸道肌肉疲劳。
断奶的审判被打断和通气支持恢复前吸气肌疲劳一旦呼吸窘迫的患者出现临床症状出现之前(在一段时间的肌肉疲劳)或许可以解释为什么有些研究人员未能文档长期膈疲劳患者未能从通风机断奶49。此外,它应该强调疲劳并不是一个“全有或全无现象”50;而收缩性的损伤更可能以连续体的形式存在。
在大多数神经肌肉疾病急性发作(如。膈麻痹或毒物,如有机磷的影响),呼吸肌肉的弱点是一种很常见的功能,并导致通气失败。弱点也可能是肌肉萎缩的结果,营养不良、电解质紊乱或不成熟的呼吸肌肉,如新生儿。在这些情况下,其余正常肌肉细胞不能产生足够的力量来维持足够了V”一个最终,他们变得疲惫。
阴险的血碳酸过多症发作
慢性高碳酸血症患者呼吸反对增加部队通过实施肺(如支气管炎和肺气肿)或胸壁(如脊柱后侧凸,极端肥胖或神经肌肉疾病),或两者兼而有之(硬皮病和多肌炎)(表2⇓)。
虽然在慢性阻塞性肺病慢性高碳酸血症是最常见的,它与一个不祥的预后相关51,导致其发生的机制并不完全理解。的关系P,有限公司2指数的气道阻塞或通气/灌注不匹配是虚弱的52,这表明非肺癌病理因素可能是有效的。
半个多世纪前,提出了两个假设来解释这些患者的慢性高碳酸血症。斯科特531920年,建议通风受损是化学因素影响呼吸的结果开车经过呼吸中心的化学环境的变化。克里斯蒂541934年,建议通气不足主要是异常呼吸力学的结果,而呼吸肌肉无法执行必要的工作提供足够的通风,因此二氧化碳水平上升。大多数hypercapnic COPD患者可以通过自愿增加实现血碳酸正常通风55使上述两个假设站不住脚的。考虑到增加V”ds/VT比发生在慢性阻塞性肺病,P,有限公司2可以保持在或接近正常水平提供了吗V”E可以保存在一个足够高的水平。这一事实,在稳态COPD患者中,深呼吸让附近的呼吸肌肉疲劳和不能容忍超过几分钟27可能表明hypercapnic COPD患者选择作为“智慧的战士”,谁,而不是增加他们的通风(一个选项,可能会导致肌肉疲劳),选择hypoventilate。
维护的问题V”E高于正常水平,这是与重大机械呼吸障碍。由于慢性阻塞性肺病患者的特点是增加气流阻力和减少动态合规,电阻和弹性负载增加,因此,吸气肌肉产生更高的力量膨胀肺。肺的气性变化导致恶性通货膨胀,这迫使吸气肌肉长度短于正常运作,减少胸内压的能力降低。更重要的是,动态的恶性通货膨胀,发展在这些患者由于呼气流量限制,对呼吸道肌肉严重拉伤,因为额外的负载,放在他们(内在呼气末正压通气(偷看我),因为操作长度和几何的障碍。
随后,之间的平衡的机械障碍呼吸和吸气肌肉的容量来应付他们,表达的P我/P我,马克斯在不利的方向转变(更多的工作/少肌肉储备)。在这种情况下,慢性阻塞性肺病患者选择减少P我通过减少VT。减少VT可能反映了降低中枢呼吸驱动,或者,或者,机械限制和/或吸气肌肉功能障碍。
低于正常的呼吸系统输出一直假设的机制在COPD患者血碳酸过多症。然而,神经驱动评估口阻塞压力被认为是慢性阻塞性肺病患者比正常人高56,尽管normocapnic之间没有显著差异被发现和hypercapnic病人。此外,神经驱动评估表面隔膜肌活动也发现增加normocapnic和hypercapnic慢性阻塞性肺病患者57。此外,自愿开车去呼吸已被证明不能减少hypercapnic慢性阻塞性肺病患者58。因此,尽管如此,在这些患者中,呼吸呼吸增加,缩短他们更好t我,这一过程的结果在一个低VT。
减少VT在很大程度上抵消了增加fR,这样V”E是保存完好。然而,快速浅呼吸不良后果。增加fR使动态恶性通货膨胀和增加V”ds/VT。呼吸的模式在慢性阻塞性肺病患者在几项研究已经检查了。比较正常的人与hypercapnic normocapnic稳定的慢性阻塞性肺病患者,Sorliet al。56指出hypercapnic病人呼吸更快和更浅比正常的人或者normocapnic主题,在平等的V”E,V”ds/VT较高的二氧化碳的家臣。减少VT和增加V”ds/VT还发现了开始和Grassino吗59在严重hypercapnic慢性阻塞性肺病患者。作者建议慢性肺泡肺换气不足可能患慢性阻塞性肺病病人吸气相结合的高负载(电阻和弹性)和更大的恶性通货膨胀,有效降低的因素P我,马克斯,从而增加开发/力量可供呼吸的分数(低效率的肌肉)。这些数据被Gorini证实et al。60,他发现VT是直接相关的t我,表明一个小VT主要是呼吸时间变化的结果。
导致改变的机制在COPD患者呼吸时间尚未明确。呼吸模式的变化可能代表行为反应,以减少呼吸困难的感觉。呼吸困难的感觉是一个复杂的感性构造,可能多因素疾病30.。研究表明,呼吸困难的感觉胸内压增加而增加需要保持气流VT,t我(相对于t合计),fR。因此,在给定V”E和呼吸力学、呼吸急促的呼吸模式决定了强度。
在研究的开始和Grassino59在慢性阻塞性肺病患者总通货膨胀(残余体积/肺活量76%)和血碳酸过多症,P我/P我,马克斯相比明显增加正常受试者(27吗与10%),一个值高度可能使肌肉疲劳4。因此这也暗示,随着病情的发展,功率输出的临界水平超过肌肉疲劳为了允许病人保持足够了V”一个。因此似乎很明显,当肌肉变得无法发展足够的力量,激活系统发挥作用,并可能改变呼吸的模式,以优化性能的肌肉和可能推迟或阻止严重的疲劳。
尽管下属机制不清楚,推测小纤维的传入刺激的繁重的工作(ergoreceptors、类型III)或有害物质(痛觉受器,IV型)修改中枢神经系统输出。
Acute-on-chronic呼吸衰竭
慢性呼吸衰竭患者急性恶化称为acute-on-chronic呼吸衰竭。患者可能会出现呼吸困难恶化,恶化后精神状态或呼吸停止相对较小,虽然经常多个,侮辱。Acute-on-chronic呼吸衰竭通常是在病人有严重的慢性阻塞性肺病。严重但稳定的慢性阻塞性肺病患者存在于一个非常关键的平衡增加的需求和有限的储备。任何潜在的因素干扰这种平衡(要求增加或减少外汇储备)会导致呼吸道肌肉疲劳和急性呼吸衰竭。
COPD患者的经验增加呼吸系统负载由于不正常的气道阻力和呼吸系统倒电容。阻力的增加是由于支气管痉挛、气道炎症或身体障碍由粘液和疤痕。弹性负载的最重要因素是动态发展时的恶性通货膨胀tE不足以让肺部缩小Vr之前下一个灵感。这往往发生在呼气流量的条件下是阻碍(增加气道阻力)或者当tE缩短(增加fR)39,61年。呼气流量等其他机制也可能推迟到期期间呼吸肌肉持续收缩。然而,最常见的动态肺恶性通货膨胀是慢性阻塞性肺病的患者中观察到展览呼气流量限制在呼吸和休息导致呼吸衰竭中发挥着重要作用。
当发生在肺容积大于呼吸Vr,一个积极的弹性反冲压力称为偷看我仍然在end-expiration。
当偷看我存在,吸气肌肉来产生更多的努力克服等量的气流开始前的压力。在这方面,偷看我作为吸气阈值会增加呼吸的静态弹性工作负载。平均来说,人们已经发现,吸入工作由于偷看我占总体的57%增加呼吸COPD病人所表现出的工作相对于正常人62年。
由于通货膨胀,潮汐呼吸发生在压力/体积曲线的陡峭的部分肺(增加倒电容),增加吸气负载。除了增加弹性载荷、动态恶性通货膨胀伴随着相应的有效性降低吸气肌肉压力发电机,因为吸气肌肉纤维变短和它们的几何排列变化(见肺恶性通货膨胀部分)。然而,acute-on-chronic呼吸衰竭患者可能显示不仅恶化的恶性通货膨胀,也其他条件导致肌无力(蛋白质/热量营养不良和类固醇肌病)。图4⇓序列的显示了一个示意图表示负责的机制,导致慢性阻塞性肺病acute-on-chronic呼吸衰竭患者。

序列的示意图表示负责的机制,导致acute-on-chronic呼吸衰竭患者的慢性阻塞性肺疾病。t合计:总呼吸周期;t我:吸气时间;te:呼气时间;R亚历山大-伍尔兹:气道阻力;EL,直流发电机:肺的动态倒电容;偷看我:内在呼气末正压通气;↓:减少;↑:增加。
这些患者的急性通气失败通常是由呼吸道感染。增加fR,它总是出现在严重慢性阻塞性肺病患者39,63年由于缩短了tE动态,进一步加剧了恶性通货膨胀,促进增加呼吸的静态弹性工作(由于偷看我和减少肺合规)。同时,急性增加气道阻力(支气管狭窄和大量的分泌物)导致增加电阻的呼吸。呼吸与此相关受损肌肉的增加工作效率会导致能源需求增加,这在一个临界点,超过减少能源可用(低氧血和膈血流受损由于强有力的收缩)和呼吸肌肉疲劳就会进一步增加P,有限公司2。
观点:肾上腺轴和通气失败
虽然过程导致急性hypercapnic呼吸衰竭已经彻底阐明,诸多病理生理学机制中负责慢性二氧化碳潴留尚未明确。慢性阻塞性肺病患者的事实很可能会增加他们的通风55选择不去,结果发展慢性高碳酸血症允许猜测它可能是“自然的智慧”,保护病人免受疾病的灾难性的后果,但血碳酸过多症的不可避免的成本。
研究并最终机制功能的多重性的部分说明“自然智慧”可能是一个有趣的和令人满意的经历调查员在这个领域。
因为强烈的应变的隔膜通常所需的足够了V”一个的条件下提高负载和/或降低效率已被证明导致呼吸肌肉的结构性破坏44,45,它可以假定一个保护机制可能存在为了保护呼吸道肌肉免受破坏。
尽管没有无可争辩的证据证明机制参与慢性二氧化碳潴留,有足够的数据,允许投机有关外围机械或化学刺激的方式传输和修改的呼吸模式36,37。
直到1970年代早期,呼吸中枢神经放电被认为是影响主要由中央和外周化学感受器和迷走神经传入纤维,并在较小程度上的肌肉传入,次要的角色,似乎。旅行感觉信息的影响通过膈神经中央呼吸中心没有很好地阐明,尽管知道nonmyelinated纤维构成整个膈神经的一大部分64年,65年。的功能存在小有髓和nonmyelinated膈传入纤维(分别类型III和IV),以及它们影响中央吸气神经活动,证实犀牛猫,它表明,这些纤维的选择性刺激影响呼吸节奏(改变t我/t合计),因此中央吸气活动36。压力的严重胸(肌肉紧张,局部缺血和积累的有毒代谢产物),传入呼吸肌肉可能发挥主导作用的特殊呼吸模式的起源。已被证明,响应特征对疲劳载荷或呼吸呼吸减少血液流动状态tachypnoea,最初其次是呼吸徐缓和呼吸停止14,37。因为这种反应不是影响动物,通过迷走神经切断术,消除迷走神经传入纤维,或cross-perfusion头,消除化学感受器传入纤维,这种建议似乎是合理的,传入的信息类型III和IV受体可能会增加他们的影响中央呼吸控制器和改变通气时机36,37。
传入膈纤维呼吸模式的影响,以及可能的刺激引发他们,一直在研究动物犀牛在平静呼吸或疲劳试验38,66年。在有意识的山羊,艰苦的电阻呼吸与两相的隔膜肌反应67年,由最初的和立即增加(促进)后跟部分减少(抑制)。反应都归因于小传入纤维激活,最初导致便利化,,在一个时间,和/或intensity-dependent方式,部分抑制通过精化β内啡肽。之间的直接联系第三和第四组传入的刺激和中央精化的内源性阿片类物质也被Kumazawa建议et al。68年表明,在犀牛狗,呼吸抑郁坚持即使撤军传入纤维刺激,而呼吸的大小抑郁症被纳洛酮显著降低。在猫和其他研究者已经证实这些结果表明,脊椎上的机制涉及的现象69年这可以通过与纳洛酮预处理32,因此强烈表明内源性阿片样物质的参与途径。
外生或内生生成的阿片类药物的作用作为一个复杂的抑制性神经递质或神经调质系统的呼吸一直得到广泛的研究。当注入池状的脑脊液、海洛因的减少造成的VT33,表明对呼吸阿片类药物的直接抑制作用。在慢性阻塞性肺病患者呼吸道反应流电阻加载缺席,纳洛酮的反应可以通过政府立即恢复34慢性气道阻力增加,这表明这些病人产生内源性阿片类物质作为一种适应性反应,为减轻长时间呼吸困难的压力。急性短期吸入电阻加载导致的减少VT意味着在unanaesthetised山羊吸气流量,以及增加水平的池状的脑脊液imunoreactiveβ内啡肽19。这两个VT和平均吸气流量可能是暂时性的增加了纳洛酮管理,证明这些影响的比例可能是冥想内源性阿片类物质的精化。这些数据暗示内源性阿片类物质引起的放电率逐步下降,尤其是呼吸系统中心,允许减少每呼吸吸气活动以减少呼吸肌肉的工作,由于减少了VT需要更少的压力发展,延缓或防止出现明显的肌肉疲劳。
虽然剧烈呼吸吸气电阻已经被发现产生β内啡肽不仅在动物身上,也在人类身上35,70年,这些物质的来源仍然是难以捉摸的,中央-肾上腺轴和外围网站等网站如脊髓和周围神经牵连其中67年。
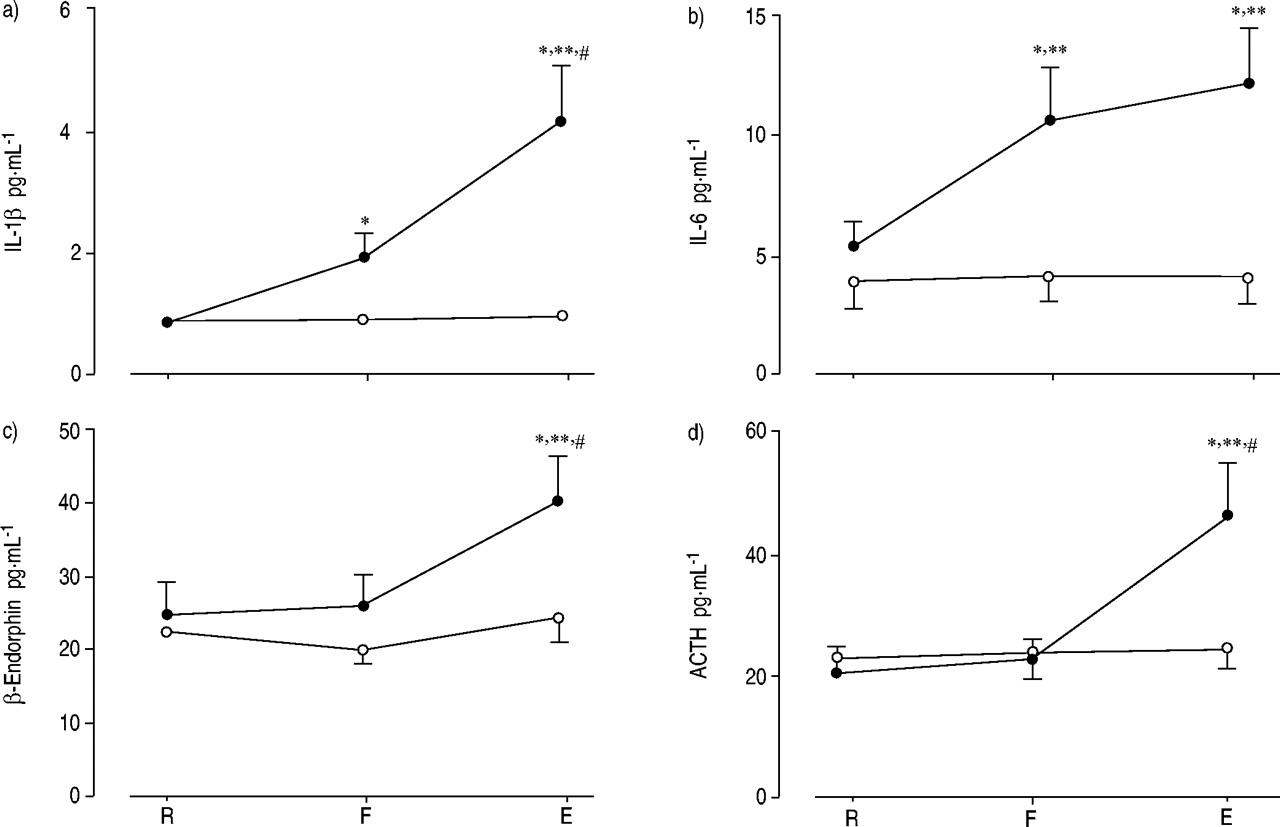
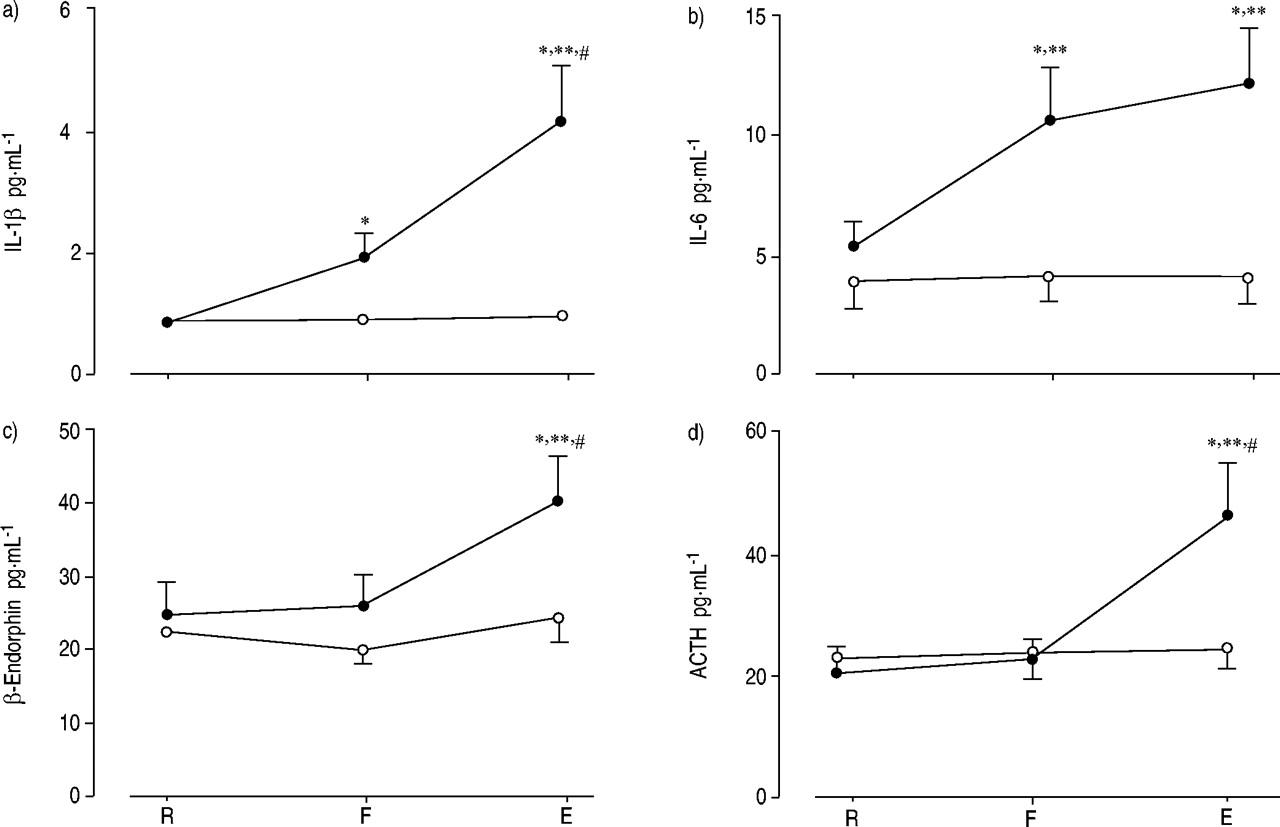
结果表明,最近,在正常人类志愿者,艰苦的电阻呼吸导致的水平显著上升的促炎细胞因子白介素(IL)高1β在IL 6、应承担的促肾上腺皮质激素(ACTH)和β内啡肽71年。之间的紧密关系β内啡肽应承担和ACTH水平上升,前循环增加IL 6水平允许作者表明,促炎细胞因子,特别是地理IL 6、负责肾上腺轴的激活,导致最终增加血浆β内啡肽和ACTH水平,考虑到都是来自同一分子的翻译修饰,pro-opiomelanocortin72年,与此同时脑垂体分泌的73年,74年。人们很容易认为机制占增加β内啡肽和ACTH水平可能是细胞因子刺激的传入神经纤维的电阻所产生的呼吸(图5所示⇓)。事实上,全球消耗的小传入纤维抑制血浆ACTH应对静脉注射IL 1β75年。

意味着等离子体水平:一个)白介素(IL) 1β;b)地理IL 6;c)β内啡肽应承担的;和d)促肾上腺皮质激素(ACTH)静止(R)的主题不能生成目标最大吸气压力(F)和电阻的最后呼吸(E)(•:高负载;○:温和的负载)。数据意味着±sem。*:p < 0.05与R;#:p < 0.05与F;* *:p < 0.01与温和的负载。(复制许可66年)。
虽然刺激生产的细胞因子尚不可知,呼吸道内活性氧产生肌肉疲劳过程中电阻的呼吸76年,77年可能负责。最近被发现,抗氧化剂政府在健康志愿者显著弱化了艰苦的电阻breathing-induced细胞因子的反应78年:肿瘤坏死因子α应承担响应被废除,IL 1β应承担成为察觉,IL 6反应迟钝。因此可能是等离子体中细胞因子诱导电阻呼吸不同受各种刺激,其中一些被普遍(活性氧),其相对重要性随每个各自的细胞因子。
尽管目前作者承认时间剖面(短暂)和强度增加的阻力(更大的)在上述模型不同于患者的慢性二氧化碳潴留,鉴于剧烈呼吸导致隔膜肌肉组成的纤维损伤膜损伤,肌节破坏44,45和乳酸生产38,下面的假设可以到达(图。6⇓)。呼吸费力的电阻,通过活性氧,导致血浆细胞因子的生产,尤其是地理IL 6,进而调节呼吸控制器可能直接通过血液或小传入或通过-肾上腺轴。随后刺激肾上腺轴的细胞因子可能有双重目的:有机体的ACTH响应可能代表试图减少伤害发生在呼吸道肌肉通过肾上腺糖皮质激素的生产79年。同时,生产β内啡肽减少呼吸肌肉的激活35和改变呼吸的模式19为了减少和/或防止进一步的伤害为代价的V”一个。
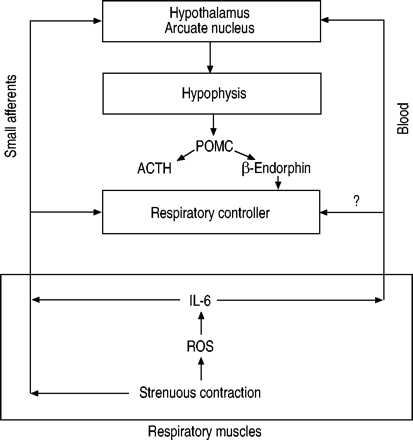
可能的循环呼吸肌肉和中央控制器之间:活性氧(ROS)、白介素(IL)高6小纤维,肾上腺轴,促肾上腺皮质激素(ACTH)和内啡肽。POMC: pro-opiomelanocortin。
结论
呼吸系统基本上由两部分组成:气体交换的器官,肺,通风肺部的泵。泵包括胸壁、呼吸肌肉取代胸壁,呼吸中枢神经系统中心控制肌肉和神经肌肉连接中心。系统的两个部分是至关重要的。一般来说,气体交换功能的失败由于肺部疾病(即。ARDS、肺炎和肺栓塞)结果主要是在低氧血与血碳酸正常或低碳酸血(I型呼吸衰竭)。泵的故障(即。CNS抑郁,缺点和创伤),也会导致低氧血,导致肺换气不足和血碳酸过多症的标志通气失败(II型呼吸衰竭)。
Hypercapnic可能发生呼吸衰竭严重,在不知不觉中或严重慢性二氧化碳潴留。在这些条件下,病理生理,公分母是减少V”一个对于一个给定的V”有限公司2。
机械障碍往往是肺泡肺换气不足的原因。障碍与肺换气不足可以很容易地在胸壁创伤(连枷胸),过度恶性通货膨胀(飞速膨大阻塞性肺疾病患者)和脊柱后侧凸。神经肌肉疾病的患者表现为虚弱,,例如,在过量,中枢神经系统损伤和神经肌肉疾病,导致肺泡肺换气不足严重或长期。能源需求和供应的不平衡可能最终会导致呼吸道肌肉疲劳(即。反过来,冲击),减少了通风导致血碳酸过多症。
广泛讨论多年没有提供答案,为什么有些患者出现慢性高碳酸血症在慢性阻塞性肺疾病,脊柱后侧凸和neuromyopathies。最具吸引力的假设在这个障碍的理论是“自然的智慧”。患者面临一个负载有两种选择:要么努力推动为了维持正常动脉氧气和二氧化碳紧张的代价最终成为疲劳和疲惫或呼吸每分通气量较低,避免呼吸困难,疲劳和疲惫但肺泡通气量的减少。信任是借给这后一个选项,目前支持作者,通过最近的工作。简单地说,有可能是一个阈值吸气负载可能存在,,只要超过,导致肌肉损伤,和,因此,一种自适应的细胞因子和激素响应是预防和/或减少引起损伤。然而,这在肺泡肺换气不足的高潮和二氧化碳潴留。
- 收到了2003年4月7日。
- 接受2003年8月7日。
- ©人期刊有限公司










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}