





文摘
三个不同的实体现在由主肺动脉克隆淋巴扩散的定义。本文的目的是描述的病理生理,这三个的诊断、预后及治疗方面的实体。
轻度肺b细胞淋巴瘤是最常见的形式的主肺动脉克隆淋巴扩散。它起源于mucosa-associated淋巴组织。通常是懒惰的和的形式出现在慢性肺泡不透明度。预后很好,但是治疗争议(简单的监控,手术或单药化疗)。高档肺b细胞淋巴瘤是罕见,通常发生在个人与一个潜在的障碍(如。免疫缺陷)。预后差,治疗选项取决于潜在的障碍。
在初选的定义包含lymphomatoid肉芽肿病肺淋巴瘤是有争议的。的克隆性增殖很少了,肺外参与频繁(上呼吸道、皮肤、肾脏、中枢神经系统,等。)。预后是非常变量,与一些作者报告完全缓解类固醇和环磷酰胺化疗和其他报告失败的组合。
淋巴瘤的扩散可能涉及肺在三个方面:1)造血的传播的非霍奇金淋巴瘤(NHL)或何杰金氏病(HD);2)通过连续的入侵从肺门或纵隔淋巴瘤的网站;和3)原发性肺参与。第一个两种情况涉及进展或复发的一个已知的淋巴瘤的障碍,和治疗的重点是血液学的障碍。第三的情况下,本文的重点,提出了一系列为pneumologists诊断和治疗问题。
原发性肺淋巴瘤(PPL)被定义为克隆淋巴扩散影响一个或两个肺(薄壁组织和/或支气管)患者没有可检测在诊断肺外参与或在随后3个月1,2。当肺肿瘤的主要网站,这个定义还包括:1)多病灶的mucosa-associated淋巴组织(麦芽)NHL;2)肺参与卫星节点(或纵隔肺门);和3)multiorgan参与lymphomatoid肉芽肿病,克隆的本质是有争议的。
人是非常罕见的。当淋巴结外侵犯形式代表NHL的24 - 50%的病例1- - - - - -3淋巴结外侵犯,PPL代表只有3 - 4%的NHL, < NHL的1%,只有-1% - 0.5的原发性肺恶性肿瘤1,4,5。当前PPL的定义1,6包括:1)低级的b细胞PPL (PPL-B),最常见的形式;2)高档PPL-B;和3)lymphomatoid肉芽肿病(LG),一种罕见的疾病。
主肺b细胞非霍奇金淋巴瘤:麦芽非霍奇金淋巴瘤
进化的概念和术语
低级PPL-B对应曾称之为“假淋巴瘤”。怀疑的任期假淋巴瘤被选中,是因为这些缓慢进行性病变的恶性性质及其相对良性的组织学方面7- - - - - -10。常规使用敏感的免疫组织化学技术5,11——和分子的基于方法17事实上表明,大多数假淋巴瘤包含克隆增殖,从而展示其淋巴瘤的本性。术语假淋巴瘤因此放弃了这些淋巴瘤被细分如下:淋巴细胞,分散的小淋巴细胞,混合分散,或与小裂细胞扩散,在制定工作;淋巴细胞、lymphoplasmocytic centrocytic centroblastic / centrocytic分散在基尔分类5,11- - - - - -;和麦芽淋巴瘤在修订后的欧美淋巴肿瘤的分类(真正的)分类18。在最新的分类(世界卫生组织(世卫组织)分类)19、麦芽淋巴瘤属于边缘带淋巴瘤,但区别节点和脾形式不同的临床行为和细胞遗传学特征19-。然而,某些情况下低级PPL-B不对应MALT-type NHL的定义和高档PPL-B也被描述1。
Mucosa-associated淋巴组织淋巴瘤
支气管mucosa-associated淋巴组织
麦芽是一个淋巴组织专注于粘膜防御1。第一次描述它胃肠道的动物模型,然后在人类回肠。它包括派尔集合淋巴结补丁,固有层,intra-epithelial淋巴细胞和肠系膜淋巴结。派尔集合淋巴结补丁淋巴结节与架构类似于淋巴结滤泡,除了边际B区高度发达的在前。固有层含有免疫球蛋白(Ig) A-secreting浆细胞和T淋巴细胞。表型,intra-epithelial淋巴细胞CD8 + t细胞。胃是最常见的麦芽淋巴瘤和作为模型肺淋巴瘤。在胃里,麦芽是缺席在生理情况下肺。在慢性抗原刺激幽门螺杆菌),麦芽可以开发的胃,进行二次起源于边缘区b细胞淋巴瘤的转换。为了发展,恶性b细胞克隆需要专门针对t细胞的存在幽门螺旋杆菌抗原。因此,幽门螺旋杆菌根除会导致冗长的完全缓解胃淋巴瘤23。没有触发抗原迄今为止发现的肺,但慢性抗原刺激某些自身免疫性疾病(系统性红斑狼疮,多发性硬化症,桥本甲状腺炎,尤其是Gougerot-Sjogren综合征)被认为是影响肺麦芽淋巴瘤的发病23。
流行病学
低级NHL-B占58 - 87%的病例病理系列PPL5,12,15,17。这些情况下对应MALT-type NHL的近90%12,15,17。发病年龄∼50 - 60岁(12 - 79岁)和主题< 30岁很少影响[5、9、11、13、14、16、24)。两性都同样影响[5、9、11、13、14、16、24)。
临床和放射的迹象
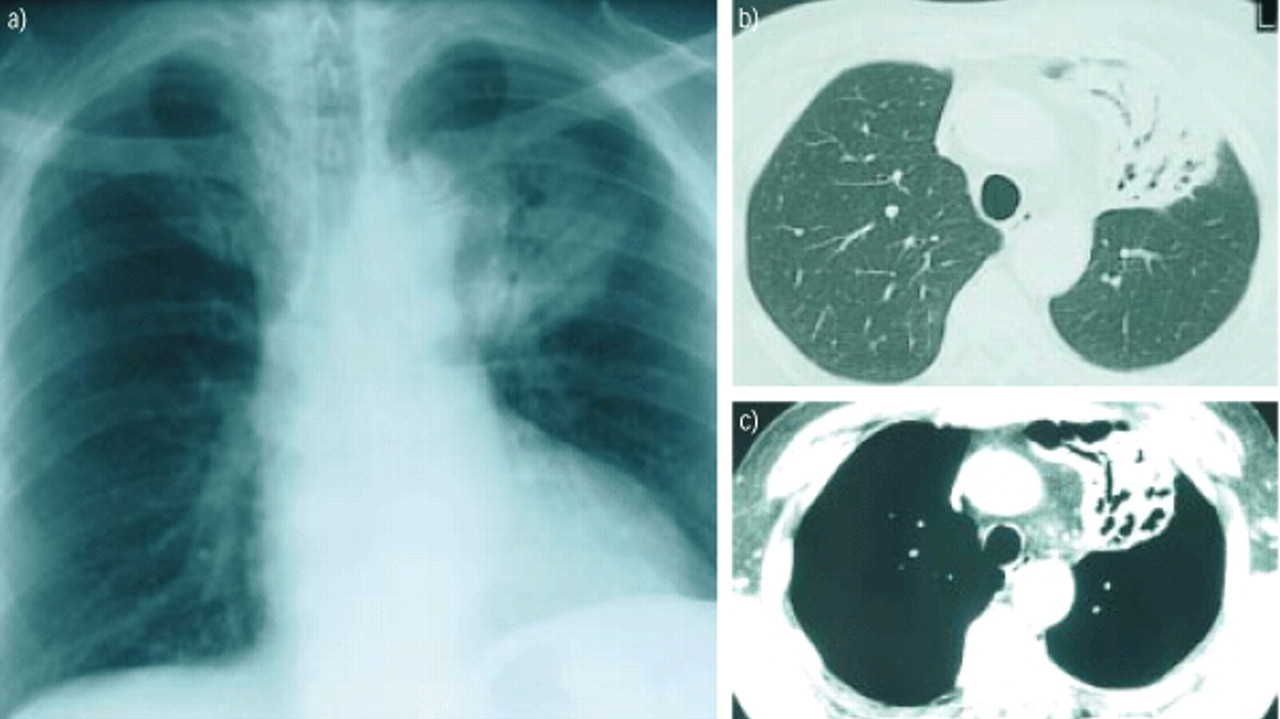
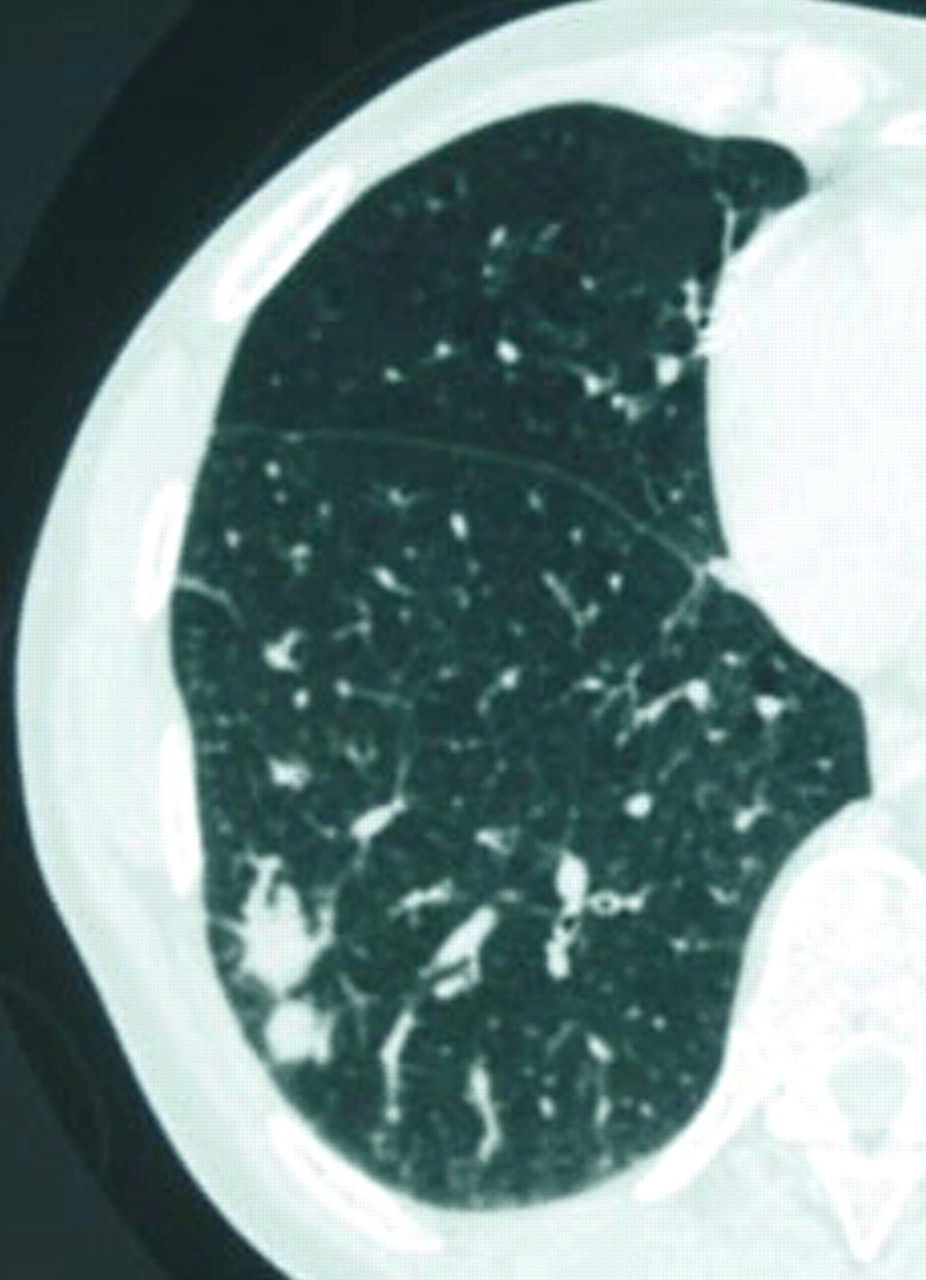
近一半患者在无症状的诊断和识别的基础上意外地放射性肺异常[24]5、11、13、14日。当礼物,症状,如咳嗽、轻度呼吸困难,偶尔胸痛,咯血,非特异性(5、9、11、13、14、16、24)。肺部听诊发现陶瓷器皿在< 20%的病例24。根据定义,肺外表现是局限于一般的迹象(发烧和减肥)和发生在不到四分之一的病人[24]5、11、13、14日。通常的辐射方面(50 - 90%的病例)是一种局部肺泡透明度,直径< 5厘米和模糊或明确的轮廓(根据系列);这是近50%的情况下与空气支气管征(图1⇓)(13、14、24)。计算机断层扫描(CT)(无花果。1 b和c⇓比标准射线照相法),这是更敏感,表明病变通常是双边(60 - 70%)和多个(70 - 77%)25,26。几乎所有这些病变包含明确的区域对应于一个完整的支气管腔(图2所示⇓)。内膨胀支气管病变的存在是一个很好的诊断标志,尽管底层机制是无法解释的26。只有不到10%的病人有双边扩散reticulonodular混浊,atelectasia或胸腔积液(13、14、24)。CT能显示肺门和纵隔腺病9,24。第一个临床或影像学表现之间的间隔和诊断范围从5个月至8年(24)5、11、13、14日。
支气管内镜的贡献
支气管内镜通常显示一个正常的宏观方面24,虽然异常从粘膜炎症到支气管狭窄可以观察到24。支气管的诊断产量,特别是transbronchial活检更高目标时可见支气管病变或影像学异常24。然而,没有具体的迹象在大多数这些样品需要进一步诊断调查[5、9、11、13、14、16、24)。支气管肺泡灌洗(BAL)是非常有用的鉴别诊断慢性肺泡透明27。相比之下,其价值正PPL的诊断评估不足。矿山已经被证明有用的人在孤立的情况下28-,它的使用并不总是提到在大型出版系列(5、9、11、13、14、16)。BAL似乎特别有价值,如果它显示淋巴细胞牙槽炎(淋巴细胞>细胞总数的20%),大约三分之二的患者中发现24,27,29日,31日。这淋巴球增多,通常主要由t细胞组成,只有一个特定的标志当> 10%的B淋巴细胞24,29日,32。b细胞牙槽炎是特别有价值的克隆时,自然可以证明了搞笑基因的检测使用的基于分子方法克隆重组(印迹或反向transcriptase-polymerase连锁反应)35- - - - - -,39,40。然而,这些方法并不是广泛应用于日常实践和需要进一步评价其敏感性和特异性。他们已经可以用于诊断为PPL的复发。筛查单克隆搞笑的球浮在表面的免疫电泳24,33,38,41和限制膜或胞浆内Ig轻链表达式通过幻灯片免疫组织化学或流式细胞术29日- - - - - -,38已经使用,但已经被分子基于其方法。
诊断标准
MALT-type NHL的诊断是基于组织学检查手术样本或支气管,transbronchial或经胸廓的活检材料。
结果常规组织学
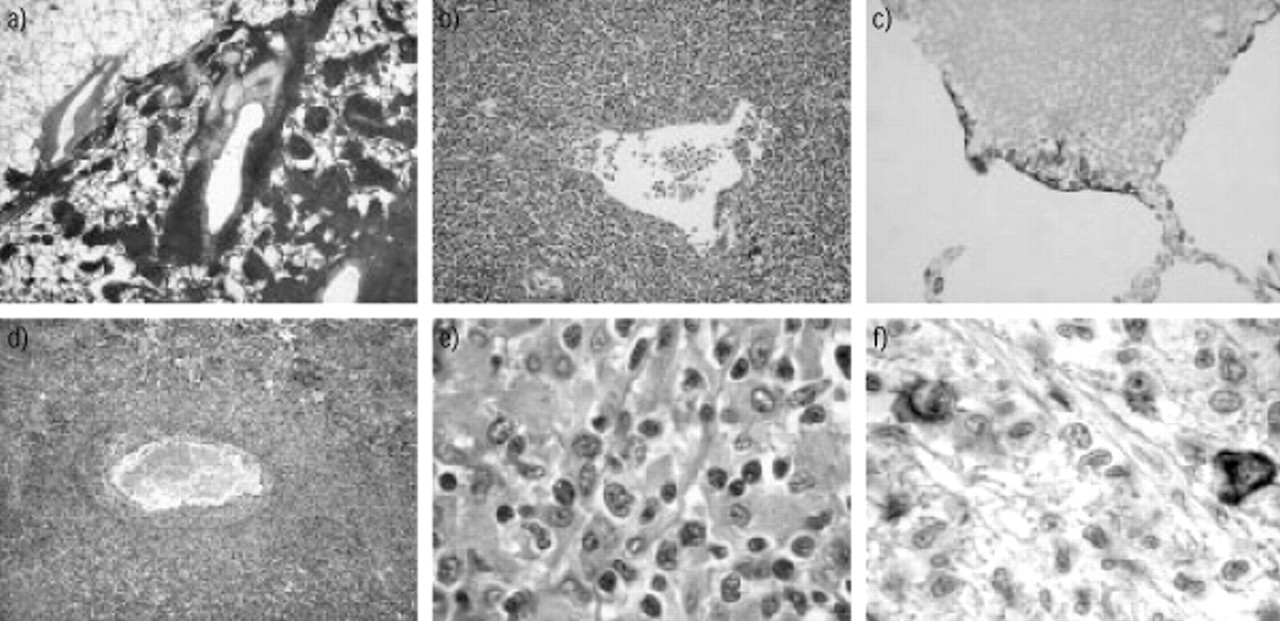
宏观方面是白色,柔软而定义糟糕的质量。显微镜下,MALT-type PPL被定义为损伤[42]11、13、15、17日,包含:1)增殖的小淋巴细胞类似于边缘区派尔集合淋巴结补丁或脾毛囊,centrocyte-like细胞和小淋巴细胞,浆细胞或monocytoid细胞;2)lymphoepithelial病变显示淋巴细胞迁移从边缘区细支气管上皮细胞;3)反应性滤泡增生;和4)罕见的再结晶的细胞。更不寻常的特性包括淀粉样蛋白沉积10,43和肉芽肿性存款10,12,43。可以发现不同程度的纤维化。淋巴瘤的渗透导致平滑或结节与支气管血管周围间质增厚分布12(图3⇓)。
免疫组织化学的贡献
免疫组织化学分析有助于MALT-type PPL的微分和积极的诊断。它显示b细胞表型(CD19 CD20)5,11自然和克隆15,17淋巴的渗透入侵滤泡结构和入侵支气管和细支气管上皮细胞(图3所示⇓)。它还揭示,持久性的树突细胞(CD21,表征CD35),存在,tumoural内扩散,破坏毛囊,在一起,在大多数情况下,用小反应性T淋巴细胞(CD3)肺泡壁渗透和周围peribronchiolar结节12,17。最重要的是,免疫组织化学测试可以排除低度恶性淋巴瘤(centrofollicular NHL-B,曼特尔NHL-B和慢性淋巴细胞白血病(CLL)类型淋巴瘤)通过展示缺乏CD5和CD10表面抗原12,17,44,45。
分子生物学的贡献
南部印迹进行冷冻样本。使用一个搞笑重链基因目标序列(Fr3 / JH),扩散的单克隆自然是12的20所示的样本MALT-type PPL17。
鉴别诊断
MALT-type PPL的鉴别诊断是根据临床和组织学依据。临床上,问题归结为诊断MALT-type PPL辐射后的识别慢性局部或弥漫性肺泡间质透明度,可以有一个广泛的目的(表1所示⇓)。
组织学检查,尤其是当样本很小,主要的困难是区分MALT-type NHL和弥漫性淋巴间质淋巴增生或肺炎(独立),和滤泡性支气管炎(神奇动物)。这种区别可能看起来无关紧要的临床医生,因为这些疾病的放射学和临床特征往往不同于MALT-type PPL47。其他组织实体,比如外在过敏性肺泡炎,可以考虑,因为他们相似的辐射表达与淋巴病变的存在。其他一些结节性肺损伤也有一个淋巴队伍,如浆细胞肉芽肿炎症pseudotumours,纤维组织细胞瘤,肺hyalinising肉芽肿、肺内的腺病,和Castleman病48。
预处理的扩展检查
淋巴瘤是排除了腹部和胸部CT对比增强。虽然在淋巴瘤骨髓参与更频繁的节点或脾边缘区21骨髓活检是至关重要的,入侵的迹象在≤20 - 30%的患者在最近一系列麦芽淋巴瘤(49,50)。同样,这些系列表明伴随其他粘膜淋巴网站参与25 - 35%的病例47——甚至更频繁的麦芽淋巴瘤患者不涉及胃肠道。寻找其他粘膜网站必须包括眼科和耳朵,鼻子和喉咙(ENT)考试(用磁共振成像(MRI)或CT的唾液和泪腺体在可疑的情况下),加上上消化道内镜(coloscopy +小肠过境,根据一些作者)。唯一有用的实验室检测的预处理检查血清电泳和免疫电泳。单克隆丙种球蛋白病(IgM的10例)被发现在20 - 60%的情况下,特别是在形式与浆细胞分化5,9,14,24。最近的一项研究表明,升高β2微球蛋白水平是穷人生存的独立预测指标50。
当然,预后和治疗
课程和预后因素
MALT-type PPL的结果通常是有利的在大多数系列的时间埋葬存活率> 80%,中位生存时间> 10年(5、9 - 11、13、15、24)。这些患者的总生存期长于的淋巴瘤患者的节点或脾边缘区21。相比之下,它尚未证明患者的生存MALT-type PPL相当于普通人群10,52。中位数生存患者胃肠道MALT-type PPL并不不同于与其他本地化的病人,但无进展生存似乎短了后者,尤其是在患者肺形式53。长期监测是必要的,由于频率的本地或extrathoracic手术切除后复发(几乎50%的病人后> 2岁)5,10,11,16,24。没有明确的麦芽淋巴瘤的预后因素已确定。在单变量分析,一些all-comer一系列麦芽淋巴瘤显示年龄超过60岁的贬义的影响,高β2微球蛋白和失败在一线治疗进入一个完整的响应50。唯一的预言者多变量分析是提高β2微球蛋白。根据其他作者,intratumoural淀粉样蛋白沉积是预后不良的因素,而lym < 1 ?显示= (fo) > phoepithelial病变预后良好52。最近一系列48 PPL确定患者没有预后因素如下:演讲中,两侧对称,肿瘤/节点/转移(TNM)阶段,手术切除,辅助化疗和一些组织学标准54。
一些作者认为低级PPL可以变换成高档扩散,基于混合形式的存在或过渡形式被连续切片5,9,12,15,17。这与最近的研究显示差异冲突低档、高档PPL的细胞遗传学异常。例如,t(11; 18)易位只是出现在低级PPL55。出于这个原因,最新修订的《世卫组织分类建议使用术语“大b细胞淋巴瘤”而不是“高档麦芽淋巴瘤”19。
治疗的原则
没有共识的治疗。缺乏一个确定的罪魁祸首抗原在肺癌、胃(相反幽门螺旋杆菌),意味着抗生素有效低级局部胃淋巴瘤是不合适的。目前的治疗方法是手术,化疗和放疗5,9- - - - - -11,14-。这些治疗方法不能分析各自的功效,然而,由于缺乏比较系列,甚至一些作者提出简单的临床监测11。然而,手术切除局部肿瘤通常是首选5,9- - - - - -11,14-。独家化疗患者通常用于双边或肺外的参与,复发或进展。组合方案,如环磷酰胺、阿霉素、oncovin和强的松(切),没有被证明比单药疗法更有效chloraminophene,环磷酰胺、硫唑嘌呤或类固醇9,11,14。放射治疗是很少使用5,9,11,14,15。
其他形式的b细胞原发性肺淋巴瘤
低级的b细胞原发性肺淋巴瘤
在< 10%的情况下,低级PPL-B MALT-type NHL的不符合组织学标准。根据世界卫生组织的分类18曼特尔,这些案件可以对应于滤泡或淋巴细胞NHL或慢性淋巴细胞白血病。临床和肺放射学方面类似于MALT-type PPL12,15。
高档主肺动脉lymphoma-B
高档NHL-B代表11 - 19%的病例的PPL发表系列12,15,17;MALT-type NHL∼50%的情况下共存。高档NHL-B通常发生在患者潜在的障碍,如实体器官(心、肺)与环孢霉素A或OKT3移植免疫抑制56,人类免疫缺陷病毒(HIV)感染60——Gougerot-Sjogren综合征64年。它让人想起高档胸膜NHL的获得性免疫缺陷综合症(AIDS)患者60,65年和胸膜后遗症患者虚脱疗法。eb病毒(EBV)与一些高档NHL发病影响肺部62年,63年,65年-。
排除感染艾滋病毒,发病年龄是∼60岁(30 - 80岁)5,9,10,15,24。患者通常症状,呼吸道症状、发烧或体重减轻5,9,10,15,24。放射性调查通常显示一个肺质量或atelectasia。胸腔积液也经常存在5,9,10,15,24。在当前作者的经验,多和/或出土的透明经常发现在感染艾滋病毒的病人61年-。支气管内窥镜检查通常是异常,出芽或渗透性的狭窄tumoural方面5,9,24。组织学诊断通常是容易,即使很小的样品(支气管,transbronchial或经胸廓的活检),由于爆炸之类的淋巴细胞的存在,并有很强的有丝分裂活动,清楚地表明恶性肿瘤5,9,24,69年。淋巴渗透侵犯支气管、血管内腔入侵或胸膜结构和坏死通常是发现。免疫母细胞和centroblastic NHL。大多数情况下是5,9,12,15,17,24。组织学有限在鉴别诊断中的作用48。高度非典型细胞学方面意味着免疫组织化学需要排除癌,黑色素瘤或肉瘤如果有一个明显的成纤维细胞的反应,特别是扩展节点淋巴瘤和白血病的参与。angiotropic病变明显提高lymphomatoid肉芽肿的可能性。
存活率较差比低级PPL中高档PPL。总的来说,平均存活时间∼8 - 10年5,9,24,但它远低于潜在障碍患者(艾滋病毒感染、移植、虚脱疗法)。进展和局部或远处复发发生之前和更频繁5,9,24。治疗后手术切除通常是基于组合用于高档节点NHL化疗方案5,9,24。
主肺plasmocytoma
主肺plasmocytoma是极其罕见的。不到50病例报告文学70年,71年。发病年龄∼40岁,尽管儿童和老年患者中描述的病例70年,71年。两性也同样受到影响。患者通常无症状,但发热、体重减轻、胸痛、呼吸困难、咳嗽,甚至咯血偶尔被描述70年,71年。最常见的放射性方面是一个孤立肺结节,但双边弥漫性肺部疾病的报道72年。一个门的腺病在< 10%的病例观察70年。支气管内镜总是正常和开胸诊断几乎总是基于结果70年。组织学病变由单纯细胞学异常的浆细胞的变化程度70年,71年。淀粉样病变可以礼物73年。根据定义,免疫组织化学研究表明单克隆胞浆内免疫球蛋白表达70年,71年,73年。区分这些情况下从低级的淋巴瘤,总是包含小淋巴细胞和反应性浆细胞表达多克隆搞笑70年,71年,73年。
疑似患者主要肺plasmocytoma骨髓瘤必须排除正常的骨髓和骨骼检查70年。治疗主要是手术切除的基础上plasmocytoma。如果手术禁忌,大多数plasmocytomas对辐射敏感的。整个2 -和时间埋葬幸存者原发性肺plasmocytoma分别为66%和40%,分别71年。尽管完整的肿瘤切除,15 - 30%的患者出现多发性骨髓瘤在几年之内70年。
肺血管内淋巴瘤
肺血管内淋巴瘤是由于非典型淋巴细胞毛细血管腔内的扩散,小动脉、小静脉和淋巴导管,很少或根本没有入侵邻薄壁组织。它通常位于中枢神经系统和皮肤。底层机制似乎某些表面受体淋巴细胞的损失,防止血管外的迁徙。肺的参与是罕见的74年,75年,约有15例文献报道。通常,临床和放射照片是弥漫性间质性肺疾病与血氧不足和发烧75年,76年。没有纵隔节点参与。有频繁的一般状态的改变和乳酸脱氢酶升高77年脑,肾或皮肤参与几乎是永远存在的74年,75年。肺动脉高血压76年和呼吸功能不全的空气滞留77年已报告。Transbronchial活检可以协助诊断77年一样,肺毛细血管血液细胞的细胞学分析78年。联合化疗方案应用于高档NHL有功效,给∼50%的完全缓解率。
Lymphomatoid肉芽肿病:immunoproliferative angiocentric肺军团
进化的概念和术语
Lymphomatoid肉芽肿病(LG)最初描述的框架内肺血管炎和肉芽肿病。它第一次被认为是一个临床和解剖实体有别于韦格纳肉芽肿病(和可能与EBV) Liebow和同事在1972年79年,80年。患者通常有一般状态的改变,肺结节和extrathoracic参与(通常是皮肤和神经)79年,80年。组织学病变是一个“非典型、angiocentric angiodestructive和肉芽肿性淋巴渗透”79年,80年。提出了几个问题因为这最初的描述:
是lymphomatoid淋巴恶性肉芽肿病?
临床上,LG的特点是没有节点,hepatosplenic或NHL骨髓参与令人回味。其自然是非常变量。自发缓解的报告34,81年,82年和长时间完全缓解在近50%的患者获得类固醇和环磷酰胺83年,84年。然而,在大多数发表系列预后是严峻的,在> 50%的病例和死亡发生,尽管结合化疗34,79年,81年,84年,85年。组织学病变有时有淋巴瘤的方面,与地区大型非典型的淋巴细胞。正宗高档NHL诊断的疾病或尸检过程中从10%升至近50%的病人。免疫组织化学和分子的基于证据显示分析的两个系列进行单克隆五11和9例的LG86年,87年。鉴于这个变量的临床过程和lesional方面,一些作者病变从1年级(最小异常)3(最大异常),基于非典型和炎性细胞的比例81年,85年,88年,89年。最近的一项研究显示相关性lesional品位和b细胞增殖指数,但不是t细胞,巨噬细胞和自然杀伤细胞增殖指数。三年级病灶的扩散指数似乎大b细胞NHL的相同90年。
Lymphomatoid肉芽肿病
流行病学
LG是罕见的。500 -已报告600例文献,案例报告的形式和一系列10 - 150个病人34,79年,82年,84年,85年,88年,99年-。发病年龄是∼30 - 50年(-85 - 2.5年)34,79年,80年,84年,85年,88年,99年-。男性似乎比女性更敏感,性别比例1 - 6.5之间的男性/女性34,79年,82年,84年,85年,88年,99年-。没有地理和种族敏感性因素被发现。
临床和放射性肺的迹象
近90%的患者在诊断和症状表现为4 - 8个月一般的历史和呼吸道症状34,79年,82年,84年,85年,88年,99年-。呼吸道症状被发现在54% > 80%的情况下,主要由咳嗽和呼吸困难。胸痛和潜在威胁生命的可能出现咯血79年,82年,99年。发烧、体重减轻或出汗发生在30 - 70%的病例。急性呼吸窘迫的报告。在一个系列中,动脉血液中氧含量平均为8.9 kPa(67毫米汞柱(40 - 100毫米汞柱)),有一个限制,阻塞性或混合综合症在40%,分别为20%和15%的情况下,34。
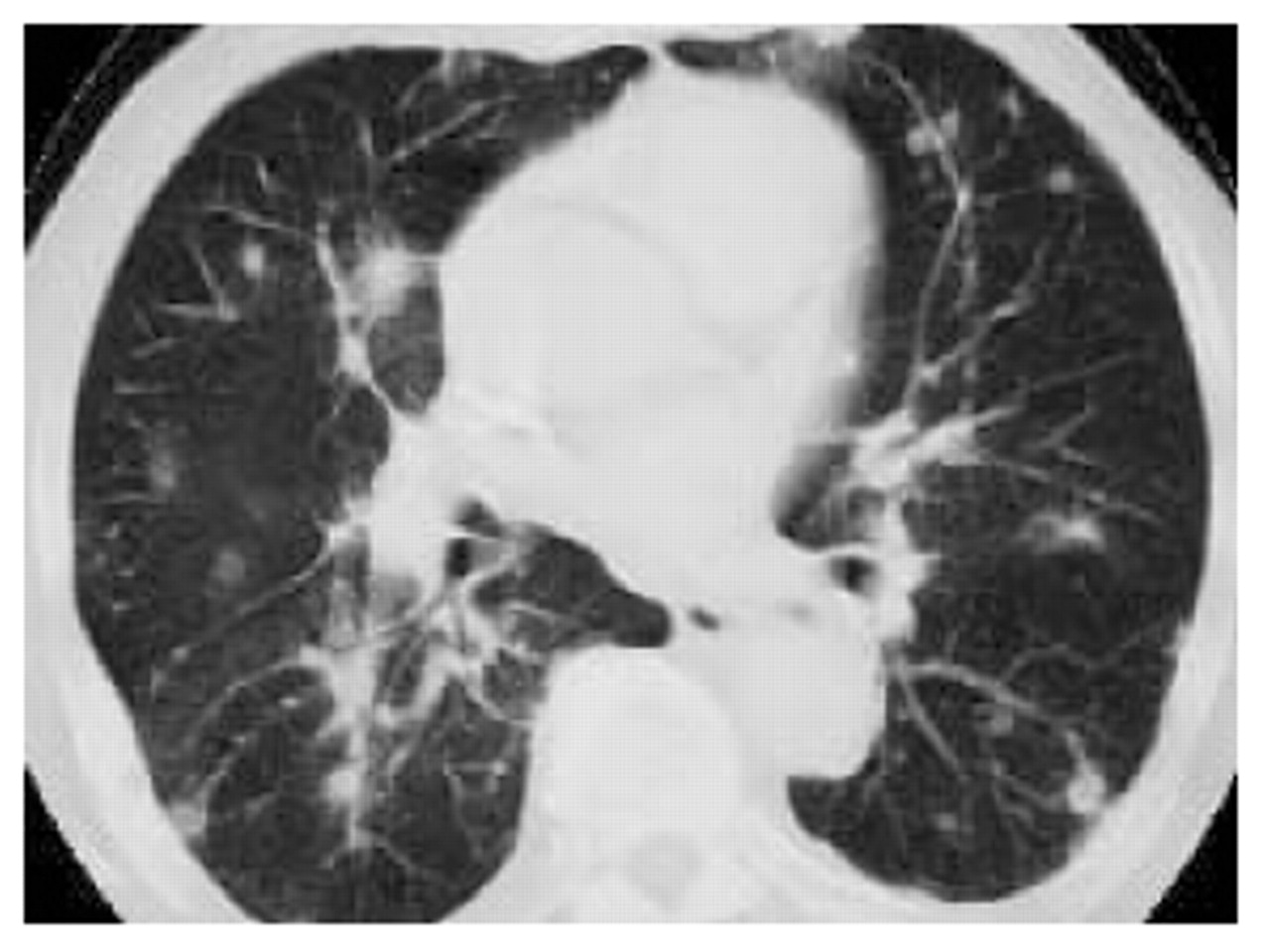
放射学方面> 80%的病例是多个定义糟糕的结节状混浊测量1 - 8厘米直径。他们是双边和低叶为主34,79年,81年,82年,84年,85年,88年,99年,102年-。结节血管分布,往往收敛形成pseudotumoural质量和挖掘,并可以消失或自发迁移(“盈亏”)82年,88年,103年(图4⇓)。Anatomical-radiological相关性研究表明,挖掘质量对应梗塞的肉芽肿病变104年,105年。单边或单个结节、肺泡间质和双边reticulonodular参与更不寻常。轻微的胸膜炎是40%的病例观察34,79年,82年,85年,99年,102年,106年和气胸患者只有被描述挖掘结节82年。门的腺病被发现在25%的情况下81年,82年,102年。
肺外表现
肺外表现主要影响皮肤、神经系统或ENT球体。他们可以之前,配合或遵循呼吸道症状的发作34,79年,81年,82年,84年,85年,88年,99年。皮肤的参与在36 - 53%的情况下,观察到,由红斑、结节,更很少,粘膜溃疡85年。神经系统参与的中央财政赤字(失明、轻偏瘫、共济失调、抽搐、昏迷,头痛,困惑,等。),影响肢体的感觉神经病变或颅神经,更很少,垂体的参与,在10 - 35%的情况下发生34,79年,81年,82年,84年,85年,88年,99年。中描述溃疡上呼吸道的10 - 30%的病例34,81年,84年,85年。肾脏病的频率是不同的报道;∼0 - 10%在疾病诊断和增加的过程中,在某些解剖系列达到了30 - 40%79年,81年,84年。它几乎总是由于质量综合症;creatinemia和尿沉积物通常是正常的。具体的外围腺病也是罕见的,在5 - 8%的情况下被观察到34,79年,81年,84年。其他肺外表现,如关节痛和眼部或胃肠道参与,观察在一些系列∼10%的病例中34,79年,81年,84年。有轶事报道许多其他器官的参与34,79年,81年,84年,85年,包括肌肉、甲状腺、肝、脾、睾丸,骨髓,肾上腺,心,前列腺癌、卵巢,等。
诊断标准
LG的诊断是基于外科肺组织学检查样品。内窥镜样本很少积极34。其他组织学活检获得的足够的样本大小有时会皮肤病变(44%的病例)积极或ENT组织(86%)34。建议检查所有访问网站,由于变量比例的非典型细胞发现在给定的时间在不同的病变。
标准的组织学分析的结果
宏观上,这些病变是相对明确的变量大小的结节,灰色、白色或淡黄色,有时坏死并形成空洞。渗透是多功能的,由淋巴细胞(小或中等大小,有时不规则,但相貌成熟染色质和核仁),与更多的不成熟的激活淋巴,plasmocytoid免疫母细胞细胞或大,或多或少的不规则,再结晶的细胞81年- - - - - -,86年,95年。细胞数量是可变的,团的细胞可以发生。有丝分裂活动也是变量(至少一个有丝分裂/低功耗领域)根据Liebowet al。79年。有丝分裂通常与周围区域坏死或血管85年。成熟浆细胞或巨噬细胞也存在。没有Sternberg细胞。多形核中性粒细胞和嗜酸性粒细胞通常缺席79年,82年,92年。同样,没有类上皮或gigantocellular肉芽肿组织79年,82年。
这种渗透影响血管的血管取向的类型(图3所示⇓),但特别是肌静脉和小动脉。细胞渗透主要是壁,提高内皮和限制内腔79年,87年。内腔入侵和血管血栓形成是少见的79年。angiocentrism有时仅染色后可见血管弹性蛋白的幻灯片,尤其是当这艘船是由一个致密细胞渗透蒙面。邻近肺组织可以港口组织肺泡损伤79年,82年。细支气管有时溃烂或被肉芽组织79年,82年。
鉴别诊断
临床诊断的主要困难是LG多个挖掘结节状的透明和患者亚急性和呼吸的迹象(表2⇓)。必须排除传染病引起局部的形式,通过组织化学染色(Ziehl格罗克特,克)。一些目的更难以识别,由于radiological-clinical和/或类似LG的组织学特性。这些目的包括韦格纳肉芽肿病,坏死性结节病和良性肉芽肿和淋巴细胞性脉管炎6,34,79年,80年,85年,88年,108年。其他组织实体也可以讨论,由于其临床表现和淋巴病变的可能性。考虑到治疗的影响,有必要排除何杰金氏病和高档angiotropic NHL诊断LG。
预处理的扩展检查
扩展检查包括寻找皮肤,特别是神经系统参与,由脑MRI。由于他们的相对频率,肾和ENT病变也应寻求。最后,节点必须寻找和骨髓的参与,因为它让LG /扩散NHL高度可能的诊断81年。
不需要特定的实验室测试诊断或预测在此设置。贫血,生物炎症综合征,白细胞增多,或者相反,白细胞减少或淋巴球减少症通常是礼物34,81年,82年,84年,85年,88年。免疫学检查显示只有非特异性异常85年,88年。抗核抗体和类风湿因子很少是积极的81年,82年。Hypogammaglobulinaemia或hypergammaglobulinaemia循环免疫复合物被描述34,81年,82年。皮肤无力回忆抗原或dinitrochlorobenze > 50%的病例中观察到84年,88年。淋巴细胞增殖抗原或有丝分裂原也是弱智者。
当然,预后和治疗
课程和预后因素
虽然“恶性肿瘤和善举”之间的界限和“monoclonality polyclonality”仍然没有得到很好的定义在LG,预后通常是可怜的。LG的患者平均存活时间是∼4岁34。在38 - 88%的情况下死亡随之而来34,79年,81年,82年,84年。患者的生存时间中位数是6.5 -19个月死亡34,81年,82年,84年,99年。死亡通常是由于窒息或咯血(44 - 89%的病例)34,79年,81年,82年,84年,99年神经系统并发症(7 - 31%的病例)或感染(23 - 38%的病例),这可能是也可能不是治疗相关34,82年,85年,99年。在许多情况下,死亡与节点相关联或内脏淋巴瘤(5 - 47%的病例)34,81年,82年,84年,85年或癌(11%)34。在幸存者中,平均生存时间是2 - 4年(≤10岁)系列中最长的后续34,81年,82年,84年,99年。生存是更好的在进入完全缓解的患者,虽然后期复发发生在∼10%的病例34,82年。
很少有研究已经确定了生存的预测因子。似乎预后良好的因素包括高龄,缺乏症状和单边放射损伤。预后不良的因素包括25岁前发病,神经病变和haepatosplenomegaly81年,82年。性别、种族和皮肤的参与不会出现影响预后至关重要。白细胞减少,持续发烧或无力出现与发展为更积极形式的LG84年。组织学检查的存在大量的大型非典型细胞和高度的坏死可能是贬义的因素81年,82年。然而,后者没有确认在最近的一项研究中,在其中所有的组织学分类与LG预测患者的生存87年。由于各种疗法用于治疗LG,它们对结果的影响很难推断生存曲线。
治疗的原则
没有共识的治疗。通常是基于类固醇治疗,单独或与环磷酰胺(CPM)相结合,并结合化疗34,79年,81年,82年,84年,99年。Fauciet al。84年获得持续完全缓解7 13例(54%)患者治疗LG的结合CPM (2 mg·公斤−1·天−1)和强的松(生产)(1毫克公斤−1·天−1,逐渐减少剂量),平均37和28个月,分别。然而,LG的8名患者(61%)死亡,包括7个相关的高档NHL抵抗化疗升级。Lipfordet al。83年也获得了完整的反应在9个(50%)患者的18 CPM和LG生产组织的成绩我(5个9),二(2 6)和三(2,3)。然而,其中9 18例(50%)随后死于高档NHL耐火材料不同的化疗方案。在本系列中,五的八个三级LG患者最初接受密集化疗结合仍然完全缓解近7年的随访时间是(1 - 12岁)。其他组也报告了类似的结果与密集的联合化疗34。这些数据表明:1)等效CPM /生产组合的有效性和更密集化疗;2)不进入缓解期的患者的预后不良;和3)对临床的需要,细胞学或遗传标准检测NHL或预测其发病。
局部肺形式已经成功接受手术或放射治疗81年,85年,88年。放射治疗也被用来治疗更加分散和/或extrathoracic形式(特别是大脑参与)23,53,82年,84年。简单的监控也可能是适当的,当自发完成缓解曾被观察到34,81年,82年。
结论
已经取得了大量的进展了解原发性肺淋巴瘤的病理生理学。巴尔病毒的作用,例如,在某些情况下现在是有据可查的高档b细胞淋巴瘤和lymphomatoid肉芽肿病。同样,它可能是一个传染病扮演了一个角色的出现肺mucosa-associated淋巴组织淋巴瘤(类似于幽门螺杆菌在胃里)。这些克隆的诊断淋巴那时也受益于免疫组织化学和分子生物学的进步。这些技术现在必须更彻底地评估在这个环境中,特别是在小内镜活检样本,以避免不必要的、纯粹的诊断胸廓切开术。这些罕见的肿瘤治疗的病人缺乏标准化和寄存器必须创建定义最好的治疗策略。
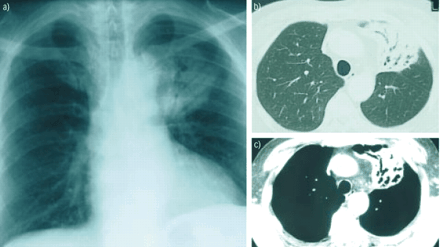
肺mucosa-associated淋巴组织(麦芽)淋巴瘤。)标准正面照相显示大量的左上叶含有空气支气管征。计算机断层扫描的部分(b)薄壁组织的和c)纵隔窗)左上叶含有空气支气管征。
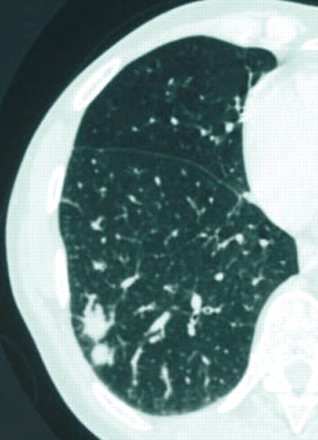
肺mucosa-associated淋巴组织(麦芽)淋巴瘤。计算机断层扫描部分(薄壁组织的窗口)显示两个加入周边结节与模糊轮廓位于右下叶。结节是bronchocentric之一。
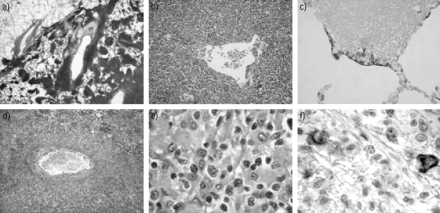
肺mucosa-associated淋巴组织(麦芽)淋巴瘤(a - c)。一)与支气管血管周围间质增生取向(他×25)。b) Lymphoepithelial病变,细支气管上皮蒙面或部分被淋巴浸润(他×200)。c)残留上皮细胞显示有一个anticytokeratin抗体(免疫组织化学,avidin-biotin过氧化物酶×100)。肺lymphomatoid肉芽肿病(d-f)。d)血管壁淋巴样增生与入侵(他×100)。e)混合细胞群与大非典型淋巴细胞和小淋巴细胞(他×1000)。f)的大型非典型淋巴细胞显示一个anti-CD20抗体(免疫组织化学,avidin-biotin过氧化物酶×1000)。
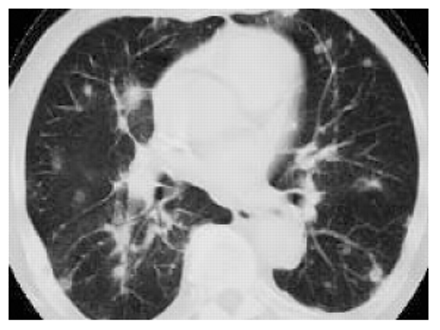
肺lymphomatoid肉芽肿病。计算机断层扫描部分(薄壁组织的窗口)显示多个结节血管分布主低叶。
脚注
↵本系列之前的文章:没有。1:钢E, Paesmans M, Berghmans T,et al。p53的角色作为生存在肺癌的预后因子:系统回顾和荟萃分析的文献。欧元和J2001;18:705 - 719。2号:范•克拉维伦RJ Habbema JDF,皮德森JH, de通力HJ Oudkerk M, Hoogsteden HC。肺癌筛查的低剂量螺旋ct。欧元和J2001;18:857 - 866。3号:Brambilla E,特拉维斯WD,科尔比电视,科林B: Shimosato y肺肿瘤的新的世界卫生组织分类。欧元和J2001;18:1059 - 1068。4号:布洛克CS,李SM。抗血管生成策略和血管靶向治疗肺癌。欧元和J2002;19日:557 - 570。5号:赫希FR,梅里克DT,富兰克林佤邦。肺癌早期诊断的生物标志物和化学预防。欧元和J2002;19日:1151 - 1158。6号:JK, Youngson JH。利物浦肺项目:早期肺癌检测的分子流行病学研究。欧元和J2002;20:464 - 479。
- 收到了2002年4月22日。
- 接受2002年4月22日。
- ©人期刊有限公司










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}