





摘要
儿童肺动脉高压(PH)中生殖系突变的患病率文献很少。本研究的目的是确定儿科队列中PH基因的突变频率,并描述突变携带者的临床特征。
本研究纳入66例PH指标:特发性肺动脉高压(IPAH)患儿35例;5例家族性多环芳烃(FPAH);肺静脉闭塞性疾病(PVOD)患儿3例;以及23名与先天性心脏病(APAH-CHD)相关的多环芳烃患儿。
23例APAH-CHD患儿未发现突变。在40名IPAH或FPAH患儿中,发现了12个突变:5个BMPR2;4ACVRL1;三下TBX4。在三个PVOD案例中,有两个携带了EIF2AK4突变。突变携带者在诊断时病情更严重,需要更积极的一线治疗。三例PVOD患者在诊断时病情非常严重,需要进行肺移植。
小儿多环芳烃的遗传结构富集于ACVRL1和TBX4突变与成人多环芳烃相比,但需要进一步的研究来证实这些结果。携带其中一种基因突变的儿童儿童期发病的多环芳烃在诊断时表现更为严重,但与非携带者观察到的结果相似。
摘要
小儿肺动脉高压具有特殊的遗传结构http://ow.ly/54yP301hCQi
介绍
肺动脉高压(PAH)是一种罕见的破坏性疾病,由血管细胞增生和内膜纤维化导致小径肺动脉进行性闭塞,导致心力衰竭,成人平均生存期<3年,儿童平均生存期<10个月[qh]1- - - - - -3.].多环芳烃可以是一种明确的病理条件的并发症,它可以发生在家族史或基因突变的背景下(遗传性多环芳烃;或者在没有确定的诱发因素的情况下被认为是特发性(IPAH) [4].IPAH病因不明,约占成人PAH的70% [5].在前瞻性TOPP(儿童PH跟踪结果和实践)登记中,收集了儿童和青少年肺动脉高压的数据,88%的儿童患有PAH,其中57%为IPAH或HPAH, 36%为PAH与先天性心脏病(APAH-CHD)相关[6].HPAH是一种遗传异质性疾病。与成人HPAH相关的主要基因是BMPR2该基因编码骨形态发生蛋白信号通路的2型受体。低外显率显性BMPR270%的家族性PAH (FPAH)和15%的IPAH中发现了突变[7,8].这些突变的外显率在两性中不同(男性为14%)与根据L阿金et al。(9])。相同途径的其他基因发生突变,ACVRL1在极少数情况下英格,在大多数情况下可引起与遗传性出血性毛细血管扩张(HHT)有关的多环芳烃[10,11].基因突变很少SMAD家族的基因,并在KCNK3和CAV1基因已被报道[12- - - - - -14].最近,TBX4突变(小髌骨综合征)已被确定与儿童期发病的多环芳烃有关[15].双等位基因隐性突变EIF2AK4基因已在遗传性肺静脉闭塞病(PVOD)和肺毛细血管瘤病的起源中发现,这两种疾病的表型密切相关,分别以静脉病变或毛细血管增生为主[16,17].
一些研究专门致力于确定儿童多环芳烃的基因突变。Harrisonet al。(18鉴定出两个BMPR2突变引起新创和两个引发hpt的突变英格和ACVRL1在16名IPAH患儿中Rosenzweiget al。(19发现;发现BMPR278例PAH患儿中有突变携带者(10%),其中大部分为FPAH(7/8)。在29例FPAH和IPAH患儿中,Pfarret al。(20.)发现BMPR24例IPAH患儿发生突变(14%),2例IPAH患儿发生突变ACVRL1和一种未知意义变异(VUS)英格.Fujiwaraet al。(21研究了21名16岁以下的多环芳烃先证者,发现了4个突变BMPR2(19%)和5个突变ACVRL1.鉴定多环芳烃基因突变对成年患者的预后有重要意义BMPR2或ACVRL1在这两组突变携带者中,突变在诊断时更年轻,死亡时间更短与非承运人(10].在小儿形式的多环芳烃,C飞驒et al。(22的发病年龄相似,但预后较差BMPR2携带者的死亡时间明显缩短,但在携带该病毒的儿童中差异不显著ACVRL1突变。
在APAH-CHD中,左向右分流与PAH之间的因果关系在大多数情况下被认为是明显的。最新的国际分类将3型APAH-CHD定义为PAH合并合并冠心病,包括小的分流缺陷,但不会导致严重的PAH,病程与IPAH相似[23].PAH基因突变在这些患者中的作用尚不清楚,在任何类型的冠心病修复术后PAH(4型)患儿中也不清楚,这些患儿继续发展为意想不到的肺血管疾病。儿科APAH-CHD患者的遗传数据有限。Pfarret al。(20.描述一个英格2型APAH-CHD(左向右分流)患者不太可能发生致病性VUSBMPR2家族性多环芳烃合并冠心病的突变Harrisonet al。(18在两例APAH-CHD中未发现任何突变。R财大et al。(24描述三BMPR23例2型APAH-CHD患儿的VUS。
考虑到最近发现的基因的调查,关于儿童期发病多环芳烃的遗传结构的数据有限。本研究旨在确定肺动脉高压(PH)基因突变在FPAH、IPAH、PVOD以及3型和4型APAH-CHD患儿中的比例,并描述其诊断时的基线临床特征及其各自的结局。
方法
研究人群
2007年1月至2013年12月,纳入66例患儿;66例无亲缘关系患者,年龄6.8±5.13岁(平均±5.13岁)sd在父母为本研究提供书面知情同意后,被纳入PH诊断的患者。所有患者均在同一儿科心脏科进行研究。在右心导管插管时确定PH诊断,并将其定义为静息时平均肺动脉压>25 mmHg,肺血管阻力指数(PVRI) >3 Wood单位·m2平均肺毛细血管楔压>12 mmHg。对所有儿童进行完整的诊断检查,以发现可能易患多环芳烃的合并症。在所有病例及其父母中搜索HHT症状。根据2013年法国尼斯商定的国际分类对患者进行分类[4].临床和生物学调查排除了所有已知的病因后,多环芳烃被认为是特发性的。当家庭中至少有两例病例报告时,病例被认为是FPAH,直至第三度亲属。1型(艾森门格综合征)和2型(左向右分流)APAH-CHD患者[23]未被纳入,因为血流动力学状态被认为足以解释多环芳烃的发生。纳入3型(n=13)和4型(n=10) APAH-CHD患者。排除畸形综合征或先天性代谢错误、已知的非整倍体(21三体)和其他细胞遗传学异常(1号和14号染色体)(n=18)。患者在诊断时进行超声心动图检查,并检测n端脑利钠肽前体(NT-proBNP)和循环内皮细胞(表1和2) [25].多环芳烃的偶发病例是在纳入研究期间诊断时进行基因分型的患者(33/40),而流行病例(7/40)在纳入研究时还活着,但经过可变延迟(7±4.5年)进行基因分型;均值±sd).同样,APAH-CHD患者既有偶发性(11/23),也有普遍性(12/23)。
对于18岁以下的儿童,建议在父母同意后对有症状的个体进行基因检测(没有父母拒绝的情况)。当发现突变时,对父母进行基因检测以区分遗传与遗传新创突变。建议所有亲属进行基因检测和详细的临床检查以诊断PH。未受影响的兄弟姐妹在父母同意后根据法国筛查未受影响儿童的法律进行基因分型。
我们的机构审查委员会和伦理委员会批准了该方案。
基因分析
所有患者都进行了筛查BMPR2和ACVRL1点突变和大的重排。IPAH或FPAH患者,且没有基因突变BMPR2或ACVRL1,进一步进行筛查EIF2AK4, TBX4和KCNK3点突变。英格和SMAD9对其他基因未发现突变的FPAH患者进行筛查(图1).自TBX4,KCNK3和EIF2AK4在研究开始时不知道与PH有关,纳入的患者对这些额外的基因进行了回顾性测试。
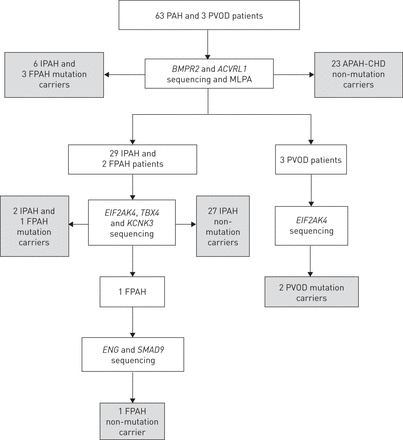
遗传分析方案流程图。每个测试的基因与用于检测突变或重排的多重连接依赖探针扩增(MLPA®)的技术一起显示。每种基因携带突变的受试者数量都被标注出来。PAH:肺动脉高压;PVOD:肺静脉闭塞性疾病;IPAH:特发性多环芳烃;家族性多环芳烃;PAH- chd:与先天性心脏病相关的PAH。
点突变和大重排的筛选按照Sztrymfet al。(7].引物用于BMPR2,ACVRL1,英格,EIF2AK4,SMAD9和KCNK3已经被描述过[7,10- - - - - -12,14,16].引物用于TBX4在补充表S1中提供了筛选。
核苷酸编号遵循人类基因组变异学会的建议。所有鉴定的突变用Alamut软件2.2版(Interactive Biosoftware, Rouen, France)进行分析。
结果
所有患者每年至少随访两次,包括世界卫生组织功能分级、NT-proBNP和超声心动图测量(表2).在分析中考虑了以下事件的发生,无论是单独发生还是合并发生:死亡;肺或心肺移植;Potts分流。
统计数据
数据以平均值±表示sd.使用XLSTAT 2014软件(Addinsoft, New York, NY, USA)进行非参数Mann-Whitney U检验,比较有或没有突变的患者,或在适当时计算卡方检验;P <0.05为差异有统计学意义。采用MedCalc统计软件12.7.7版(MedCalc software bvba, Ostend, Belgium)构建Kaplan-Meier生存曲线(图2)并计算log-rank检验,该检验用于检验突变携带者和非携带者(仅限事件病例)在三种主要事件(死亡、移植、Potts分流)中的自由概率分布的差异。
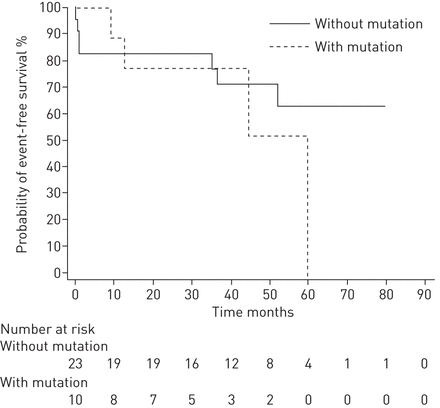
肺动脉高压患者突变携带者状态的无事件生存概率分布(仅限事件病例)。考虑的事件:死亡;移植;Potts分流。采用log-rank检验比较两种分布曲线:χ 2 =0.6680;d.f = 1;p = 0.4137。
结果
PH基因突变
经系谱分析和临床检查,35例患儿为IPAH, 5例为FPAH, 13例为3型APAH-CHD, 10例为4型APAH-CHD, 3例为PVOD。66名儿童中有14名发现了突变:8名IPAH患者(占IPAH的23%),4名FPAH患者(占FPAH的80%),2名PVOD患者(占PVOD的67%)。没有突变BMPR2和ACVRL13型和4型APAH-CHD患儿(表3).
五个BMPR2(IPAH三个,FPAH两个),四个ACVRL1(三个IPAH和一个FPAH),三个TBX42例IPAH, 1例FPAH, 2例双等位基因EIF2AK4发现了两个PVOD突变(表3).
有十种突变先前被描述并归类为有害突变。发现了四个新的突变:一个大的重排BMPR2(2-7外显子的缺失)和3TBX4所有的突变都是非义突变(所有的突变都在补充表S2中描述)。
未发现基因突变ENG, KCNK3或SMAD9基因。这四个ACVRL1突变携带者表现出HHT症状在鉴定出突变的14例指标病例中,对11例进行亲本遗传筛查。在两个IPAH病例中BMPR2鉴定出的突变有新创突变。其余的突变是遗传的,在所有情况下,携带突变的亲本无PAH症状,或携带单个等位基因的PAHEIF2AK4PVOD的突变。
在这三个家庭里TBX4突变,突变是遗传的。携带突变的父母无肺动脉高压症状,超声心动图显示肺动脉压(PAP)正常,但与骨异常相关的症状TBX4突变。
基因突变EIF2AK4基因在两例患者中被鉴定出来(先前在Eyryet al。(16]),表现出PVOD的临床、血流动力学和胸部计算机断层扫描(CT)特征EIF2AK4在IPAH或FPAH患儿中发现了突变。一名8岁女孩在诊断时胸部CT上怀疑有PVOD,但没有突变EIF2AK4在其他基因测试中也没有发现。随后的CT扫描,肺活检和进化证实了诊断,在随访期间,她的临床状况需要移植。
IPAH和FPAH患者诊断时的临床状况
在诊断时,PAH基因突变的IPAH和FPAH患者处于较差的功能等级,有更频繁的相关右室功能障碍迹象(高右房压)和更频繁的晕厥(表1).虽然在统计学上,他们的平均PAP、PVRI和心脏指数没有差异,但这些参数的值在突变携带者中往往更差。只有一个TBX4根据Sitbon标准,突变携带者在急性血管舒张试验中被认为是有反应的[26].因此,在该组中,前期联合治疗(口服内皮素受体拮抗剂和磷酸二酯酶5型抑制剂联合治疗)或前列腺素类似物三联治疗更常被用作一线治疗。值得注意的是,在成年人群中观察到女性优势,但突变携带者和非携带者的性别比例相似。两组在诊断年龄上无差异。
两组诊断时更严重的临床和血流动力学状况在2岁以下儿童组中被过度代表(4例突变,8例无突变)。
表2显示每种突变类型患者的基线特征。
讨论
本研究观察了一组患有各种形式PH的儿童的遗传状况,以更好地确定儿童多环芳烃的遗传结构,并评估与不同基因突变相关的临床状况。纳入IPAH、FPAH、PVOD以及3型和4型APAH-CHD患者,筛选已知PAH和PVOD基因突变,包括最近发现的基因(TBX4,KCNK3和EIF2AK4).多环芳烃是全身性疾病的一部分或与染色体异常相关的患者未包括在研究中。
与冠心病合并的PAH(3型apah -冠心病)和术后PAH(4型apah -冠心病)均未发生突变BMPR2而在ACVRL1提示其遗传背景与IPAH不同。在这两组中,没有易感突变表明,要么是血流动力学因素足以诱发PAH,要么是其他尚未确定的遗传因素参与了这种情况。如果遗传背景相同,这种规模的样本有94%的能力(数据未显示)检测到被测基因中的任何易感性变异。先前发表的关于一小部分APAH-CHD患者的研究结果没有检测到两者的致病性突变BMPR2或ACVRL1基因;只有误感VUS(大多数可能是良性的)在网上工具)被发现,但没有有害的突变,在相同的背景下[18,20.,24].因此,这项研究的结果和已经发表的结果都不能证明用这项研究中使用的基因筛选这些患者是合理的。
在FPAH,BMPR2在不到一半的索引病例中发现了突变,而BMPR2在超过80%的成人家族病例中发现了突变[27和其他基因突变ACVRL1和TBX4这代表了在儿童中发现的近50%的突变。同样,在IPAH中,在BMPR2在成年多环芳烃人群中发现了相似比例的突变,突变也在ACVRL1和TBX4基因的频率与BMPR2如果把突变放在一起考虑的话。由于样本量较小,本研究缺乏统计能力,无法得出成年FPAH患者突变分布的显著差异;需要进一步的研究来证实这些差异。ACVRL1本研究中发现的错义突变已经在HHT患者中有报道,并且在有HHT症状的儿童或携带HHT的父母的家庭中也有发现,因为多环芳烃可以在幼儿出现其他HHT症状之前出现,但通常出现在携带HHT的父母身上[10].事实上,克irerdet al。(10先前的研究表明,在成人中,多环芳烃发病的年龄与基因突变有关ACVRL1早于BMPR2突变携带者和这个发病年龄比没有检测到突变的成年人早。
TBX4在我们的研究中,IPAH/FPAH患者中有3/40(7.5%)检测到突变,这一比例低于Kerstjens -Frederikseet al。(15),TBX4在无发育异常的多环芳烃患儿中,有21%检测到突变。在儿童和成人多环芳烃中对该基因进行常规检测将有助于确定其重要性TBX4.
TBX4突变遗传自父母,表现为典型的小髌骨综合征的骨骼畸形,但没有多环芳烃的症状。的确,基因突变TBX4基因很少引起成人多环芳烃(根据Kerstjens -Frederikseet al。(15])。
目前,肺外显率的主要变化是TBX4从一代到下一代的突变以及在一些儿童中观察到的多环芳烃的严重程度无法解释。与来自父母一方遗传的遗传因素的相互作用和/或参与表观遗传机制是可能的假设,需要进一步研究。无成人受多环芳烃影响及携带TBX4突变可能是由于这种儿童期发病的多环芳烃的高死亡率,但目前没有随访和家庭数据来支持这种假设。我们在常规搜索中有限的经验(n=73)TBX4成年多环芳烃患者的突变频率与已报道的相似(数据未显示)[15].
所有三名患有PVOD的儿童都有非常迅速的临床恶化并进行了肺移植。虽然这三名患者有相似的进化导致肺移植,但没有突变EIF2AK4基因是在三名没有患病兄弟姐妹的患者中的一名身上发现的。研究中未发现遗传异质性yryet al。(16],但其他未知的遗传因素可能与该患者有关。
在这项研究中,突变携带者在诊断时有更严重的临床表现,这与先前报道的成年患者的经验一致BMPR2和ACVRL1突变(7,10].然而,在血流动力学状态方面的差异没有达到显著性,可能是由于患者数量较少。由于诊断时的临床表现较差,该组的初始治疗更为积极(表2),但这种差异在结果(表4).有趣的是,在成人和儿童多环芳烃中,BMPR2突变携带者对钙通道阻滞剂几乎没有反应。在本研究的一系列儿童期发病的多环芳烃中,除了一名患有aTBX4突变(表2和3.) [19,28,29].
在这项研究中,突变的识别并不影响初始治疗;这是基于在获得基因筛选结果之前的通常标准。如果排除所有病例都需要肺移植的PVOD患者,情况也是如此。
在成人中,BMPR2和ACVRL1突变携带者在诊断时也有更严重的症状,比非突变携带者死得更早[7,10,29].本研究的样本量不允许根据突变携带者的状态来检测结果的任何显著差异(表4和图2)疾病表现较严重,诊断后立即需要肺移植或Potts吻合的患者为PVOD患者(EIF2AK4突变)或ACVRL1突变携带者也见于成人[10].在Kerstjens -Frederikseet al。(15,死亡率没有增加TBX4与其他儿童期发病的PAH患者相比这项研究也是如此。
本研究有一定的局限性,因为它涉及的是一种罕见的疾病,而且样本量很小。权力的缺乏或许可以解释群体之间没有显著差异的原因。允许可被视为幸存者的流行病例在纳入期内出现,可能会改变突变携带者的频率。然而,突变载体频率在流行人群中相似(2/7;29%)和意外病例(10/33;30%)。这与APAH无关,因为在整个组中未检测到突变。虽然不太可能,但一些突变可能被遗漏了,因为一些患者没有被包括在内,或者因为一些基因没有在研究的所有患者中被筛选(图1).
综上所述,本研究表明筛查3型和4型APAH-CHD患者BMPR2和ACVRL1是不利的,这一结果证实了在其他研究中没有发现有害突变[18,20.,24].多环芳烃患者突变的比例与成人相当,但相关基因的分布非常不同。TBX4突变的重要贡献指出,这些突变的外显率在成人和儿童之间是不同的。
最后,突变患者在诊断时更严重的临床状况似乎与之前在成年人群中观察到的情况相当。
本研究的系列不允许对突变儿童人群的初始治疗提出具体建议。在我们的研究中,一个基因突变的存在并不能预测临床结果EIF2AK4基因。在大量儿童IPAH和FPAH患者中筛查这些突变可能有助于进一步确定这些突变对临床结果的预测价值。
儿科多环芳烃遗传诊断的一个关键问题是检测亲属携带或非携带状态的可能性,例如PVOD作为隐性疾病传播的情况下的兄弟姐妹。早期诊断和仔细随访的好处应超过在症状出现之前就对严重疾病进行诊断的损害,因为症状出现的可能性尚未精确确定。了解某些形式的儿科高血压预后不良将是父母高度情绪压力的一个来源,但这将有助于他们做出父母的决定。在这些情况下,由遗传学家和遗传咨询师提供的遗传咨询应该帮助父母清楚地了解遗传情况,并为他们的决策提供信息。
致谢
我们感谢Anne Leroy, Marie-Christine Waill和Salim Bakas,遗传学部门,GH Pitié-Salpêtrière,巴黎公共援助Hôpitaux,巴黎,法国巴黎,对患者进行基因分型。我们感谢David Tregouët, INSERM,心脏代谢和营养研究所,UMR_S 1166-ICAN,巴黎,法国,对统计数据分析的建议,以及David Smadja教授,血液学系,AP-HP, Hôpital europ
作者贡献:M. Levy, D. Bonnet:患者临床护理,研究设计,文章写作。I. Szezepanski:病人护理,数据收集。F. Soubrier, M. Eyries:基因数据,研究设计,文章写作。S. Nadaud, M. Ladouceur:数据统计分析,文章评论。
脚注
编辑评论Eur呼吸器2016;48: 987 - 989。
本文的补充资料来自www.qdcxjkg.com
利益冲突:与本文一起披露的信息可在以下网址找到www.qdcxjkg.com
- 收到了2016年1月28日。
- 接受2016年6月7日。
- 版权所有©ERS 2016











{kind=link}
{kind=link}
{kind=link}
{kind=link}