





文摘
肺动脉高血压的遗传原因(多环芳烃)和肺静脉阻塞疾病(PVOD)已确定,导致越来越多的遗传咨询的必要性。
在2003年至2014年之间,遗传咨询提供529多环芳烃和100 PVOD病人在法国转诊中心肺动脉高压。
PAH-predisposing突变基因被确定在72年病人的多环芳烃(17%的病例;62年的突变BMPR2,9ACVRL1(ALK1),另一个在英格)和94年PAH患者家族史(89%的病例;89年的突变BMPR2,三个ACVRL1(ALK1)和两个KCNK3)。Bi-allelic突变EIF2AK4被确定在所有患者的家族史PVOD (n = 19)和七个病人(8.6%)呈现为零星PVOD。Pre-symptomatic遗传的基因诊断提供给272亲属PAH患者,确定突变在36.4%。筛查项目是现在提供给无症状突变携带者检测多环芳烃在早期阶段,在无症状的识别结果的预测因子BMPR2突变携带者。BMPR2筛选允许我们提供两对夫妇的胚胎植入前诊断BMPR2突变。
遗传咨询可以实现在肺动脉高压中心。
文摘
遗传的原因多环芳烃和PVOD已确定;遗传咨询可以实现在PH值中心http://ow.ly/TLbee
介绍
肺动脉高压(PAH)是一种罕见的和严重的疾病影响小的肺动脉,异常增殖的肺动脉平滑肌细胞和内皮细胞导致肺血管阻力增加和右心室衰竭(1- - - - - -3]。多环芳烃可能发生在许多不同的临床上下文等结缔组织疾病,艾滋病毒感染、先天性心脏病、门脉高压和anorexigen接触(4]。零星的疾病发生时没有任何家族病史或诱发条件,多环芳烃被称为特发性。1954年,维resdaleet al。(5]描述的第一个病例多环芳烃发生在家庭环境中,提出一种遗传疾病的存在。从那时起,许多家族PAH病例描述,允许一个理解遗传PAH传播不完全外显率的常染色体显性遗传特点。虽然遗传期待以前建议发生可遗传的多环芳烃,这是最近展示了一个人工制品与不完整的观察时间家族PAH-predisposing突变基因携带者(6]。
2000年,突变的BMPR2(骨形态形成蛋白受体II型)基因的遗传多环芳烃的主要原因。这个基因编码一个II型受体(BMPRII)属于总科的转化生长因子(TGF) -β[7- - - - - -10]。BMPRII受体最初描述参与骨骼和软骨的生长和分化的规定(11,12),但最近,这也是证明中发挥关键作用的规定增长、分化和凋亡的细胞类型,包括内皮细胞和肺动脉平滑肌细胞(13]。多环芳烃发生时在家庭环境下,在种系突变BMPR2检测到至少70%的情况下(14- - - - - -19]。BMPR2突变也可以发现在3.5显然零星的PAH病例的-40% (14- - - - - -16,20.]。PAH的发生在个人或家族病史的患者遗传性出血性毛细血管扩张(HHT)允许其他两个基因遗传性出血性毛细血管扩张症发展的多环芳烃和确认:ACVRL1(ALK1)(激活素受体类型II-like激酶1)英格(endoglin) [16,21])。罕见突变Smad基因编码的蛋白质参与TGF-β信号通路也被报道(SMAD9(22,23),而SMAD1和SMAD4(22])。2012年,一个贾斯廷等。(24)发现的参与CAV-1(caveolin-1)在PAH突变,使用whole-exome测序与多环芳烃研究第三代multiple-affected家庭成员。2013年,米一个等。(25]演示的参与channelopathy KCNK3(钾通道,两个孔隙域亚K, 3)成员发展的遗传多环芳烃,确定三个家族PAH患者和三个特发性肺动脉高压病人携带的突变KCNK3基因。
肺部静脉阻塞疾病(PVOD)是一种罕见的疾病,可误诊为PAH (26),但肺血管重塑主要见于肺小静脉和小静脉PVOD [27,28]。最近,我们已经证明了的参与bi-allelic突变EIF2AK4(真核翻译起始因子2α激酶4)发展的基因遗传PVOD [19,29日]。PVOD由于EIF2AK4突变是一个未知的外显率的常染色体隐性疾病。罕见PVOD病例报告的病人BMPR2突变(27,28),但仔细的临床和病理新创评估这些情况应该承担,一起寻找EIF2AK4突变。
自2003年1月,法国转诊中心肺动脉高压提供遗传咨询和检测所有PAH患者认为是零星的,家族性或诱导anorexigen曝光,零星的和家族PVOD病人。在这里,我们审查12年经验遗传咨询的肺动脉高压。
材料和方法
病人
多环芳烃被定义为一个平均肺动脉压(肺动脉平均)≥25毫米汞柱与正常肺动脉楔压(≤15毫米汞柱)测量时对心脏catheterisation后其他pre-capillary肺动脉高压的原因已经消除。血液动力学的心脏catheterisation进行正确评价的基准在所有科目根据我们先前描述的协议(30.,31日]。后被诊断为特发性多环芳烃排除相关条件和缺乏PAH的家族史,总结在更新分类系统(4]。家族PAH承认如果有不止一个确诊病例的PAH的家庭。
PVOD被视为确认存在广泛而分散肺静脉和小静脉梗阻肺活检,事后肺癌或肺移植后肺得到样品。PVOD被认为是“非常可能”当病人满足以下特点:pre-capillary肺动脉高压,存在两个或两个以上的放射性异常特征PVOD在胸部的高分辨率计算机断层扫描(淋巴结肿大、小叶中心的毛玻璃的透明和小叶间隔线)和较低的扩散能力的肺一氧化碳血红蛋白水平纠正(DLCOc)。的诊断PVOD也支持的特定的多环芳烃治疗开始后的肺部水肿的发展(27,31日,32]。
法国对遗传咨询的建议
生物伦理学遗传咨询遵循法律》(2008 - 321年的4月4日,2008)规定的条件出发,进行评论一个人的遗传特性及其遗传指纹鉴定医学目的和修改代码(公共卫生表1)。
法国的道德原则指导遗传咨询和2015年联合欧洲呼吸学会(ERS)和欧洲心脏病学会(ESC)肺动脉高压的诊断和治疗指南188bet官网地址1,2)告知患者正确,以避免伤害,让病人保持自治(披露这一过程中,风险和收益的基因测试没有外部压力),并允许平等获取遗传咨询和检测。我们鼓励病人与家人分享遗传信息。咨询心理学家是系统提供给所有的病人和亲属,因为遗传的心理影响的结果。
迄今为止,预测基因检测对18岁以下儿童仍是一个有争议的问题,因为缺乏预防和治疗治疗的多环芳烃。因此,在法国,没有基因检测多环芳烃目前提供给18岁以下儿童无症状。
基因检测在法国转诊中心肺动脉高压
2003年1月以来,法国转诊中心提供遗传咨询和肺动脉高压BMPR2突变筛查(点突变和重组大)所有PAH患者认为是特发性,PAH家族史的患者,患者暴露于anorexigen药物和肺动脉高压患者与其他罕见的疾病有关。当没有突变BMPR2被发现在PAH患者家族史,筛选吗ACVRL1(ALK1),英格执行(点突变和大型重组)。作为ACVRL1(ALK1),英格遗传性出血性毛细血管扩张症突变与成套外显率只在一个先进的年龄、临床症状的遗传性出血性毛细血管扩张症是寻求在所有患者在遗传咨询和筛查ACVRL1(ALK1)突变进行35岁以下的病人或者遗传性出血性毛细血管扩张症的临床体征在场(复发epistaxes,黏膜与皮肤的telangiectases和动静脉畸形,特别是在肺,肝,脑循环)。如果没有突变BMPR2,ACVRL1(ALK1),英格被确定在PAH患者家族史,筛选吗BMPR2启动子,SMAD9,KCNK3和CAV-1随后进行的基因。
我们最近演示了bi-allelic的参与EIF2AK4突变的主要原因遗传PVOD [29日]。自2014年以来,病人是在我们中心(普遍或事件)确认或“非常可能”PVOD还测试了EIF2AK4突变。如果没有EIF2AK4突变被发现在一个病人显示“非常可能”PVOD或家族史的PAH / PVOD PAH-predisposing基因筛查。所有患者进行基因测试前提供书面知情同意。这些程序遵循最近2015 ECS /人指南(1,2]。
分子的方法
点突变筛查的编码区域和intronic连接BMPR2,ACVRL1(ALK1),英格,SMAD9,KCNK3,CAV-1和EIF2AK4
人类基因组DNA是遗传样品准备使用semi-automatised Extragene®器(基因组行业、Archamps、法国)和DNA提取工具包(Charbonnieres-les-Bains Promega法国,法国)根据制造商的标准协议或DNA提取血液通过QIAmp列提取DNA工具包(试剂盒、Courtaboeuf、法国)。所有编码外显子和intronic连接BMPR2 ACVRL1 (ALK1), ENG SMAD9, KCNK3 EIF2AK4和CAV-1intronic侧翼放大了PCR引物,必要时附加的其实引物。2066 - bp序列起始密码子的上游BMPR2被分成五个片段的放大BMPR2启动子(所有可用PCR引物细节要求)。
扩增产品菌进行纯化和直接测序如前所述16]。由此产生的序列数据分析与SeqScape version 2.5(美国应用生物系统公司,培育城市,CA)相比,基因库参考序列BMPR2(NM_001204.6),ACVRL1(ALK1)(NM_000020.2),英格(NM_001114753.1),SMAD9(NM_005905.5),KCNK3(NM_002246),EIF2AK4(NM_001013703)和CAV-1(NM_001753)。在所有情况下,任何序列变异发现确认两次观察到的变化决定的。核苷酸编号遵循cDNA编号+ 1对应ATG翻译起始密码子的参考序列(www.hgvs.org/mutnomen)。
筛选大重组BMPR2,ACVRL1(ALK1),英格
莎莎多元ligation-dependent探测放大P093遗传性出血性毛细血管扩张症probemix工具包(MRC-Holland,阿姆斯特丹,荷兰)被用来屏幕的一个或多个外显子的重组BMPR2,ACVRL1(ALK1),英格根据制造商的标准协议。片段的分析进行多重PCR ABI 3730 DNA分析仪和结果分析使用GeneMapper version 4.0(美国应用生物系统公司,培育城市,CA)。两个DNA样本影响个人被用作控制在每一个实验。DNA电泳图叠加在这些生成的控制通过调整峰值相同的水平作为控制扩增子的获得。
结果
PAH患者的遗传咨询和检测
2014年12月,遗传咨询和检测提供529 PAH患者,其中包括来自77个家庭的106名患者的家族史多环芳烃。在这个组,492例(93%)患者在PAH超过18岁。PAH-predisposing突变基因被确定在17% (n = 72)的患者认为特发性或anorexigen-induced疾病。在这个人群中,我们确定了62 (86%)BMPR2突变携带者,9 (13%)ACVRL1(ALK1)突变携带者和一个(1%)英格突变载体(图1一个)。PAH-predisposing突变基因被确定在89% (n = 94)的家族PAH病例。其中,89年进行BMPR2突变,三个携带一个ACVRL1(ALK1)突变和两个了KCNK3突变(图1 b)。突变多环芳烃的分布提出了图2和在线补充表中列出了所有突变确定S1。
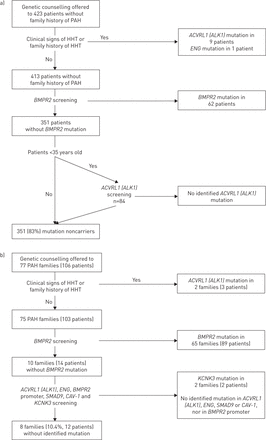
)肺动脉高血压筛查(PAH)诱发基因PAH患者一个没有家庭的环境中多环芳烃的历史。b)筛选PAH-predisposing PAH基因的家庭。遗传性出血性毛细血管扩张症:遗传性出血性毛细血管扩张。
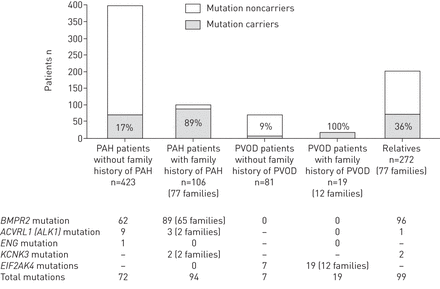
突变分布零星,家族性肺动脉高压(PAH)和肺静脉阻塞疾病(PVOD)患者和无症状的亲戚。-:未测试。
641年提出的遗传咨询和检测的实例,3例患者拒绝遗传咨询和九拒绝遗传咨询后的基因检测,占< 2%的病人。病人的原因包括无力应对合规的阳性结果还是因为宗教信仰对基因的方法。
BMPR2在PAH突变发现病人
我们确定了127种不同的BMPR2突变(在线补充表S1)。这些突变传遍BMPR2除了外显子13 (图3)。我们确定了29个错义突变,59无意义突变,13个接头缺陷,23个删除一个重复,两个变量在启动子区域。
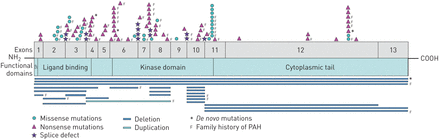
本地化的BMPR2突变中确定肺动脉高血压(PAH)患者随后在法国转诊中心肺动脉高压。SP:信号肽。
虽然胞质尾域代表BMPRII蛋白质的大小的50%,87年的101点突变(86%)局部的激酶结构域或BMPRII蛋白质和细胞外领域的只有14个点突变(14%)在胞质尾局部域(33]。
为了估计的频率新创突变BMPR2,我们选择了PAH患者携带的子群BMPR2突变但没有PAH的家族史。在这些病人中,我们寻找家族的存在BMPR2从父母的突变的DNA样本。DNA样本可以从父母双方在13个PAH患者满足上述标准。在4名患者中,家族BMPR2突变并不是发现在父母和不正确的亲子鉴定的可能性被排除的分析信息标记(图4)。三个四个病人(ABH058 ABH100和ABH177)是一个突变的携带者之前确认在我们的群(图3和在线补充表S2)。突变c。449dup identified in patient ABH086 was a unique mutation not identified in any other patients in our cohort. Clinical and haemodynamic characteristics at baseline of these four patients are presented in online supplementary table S2 and were similar to other heritable PAH cases [16,34]。
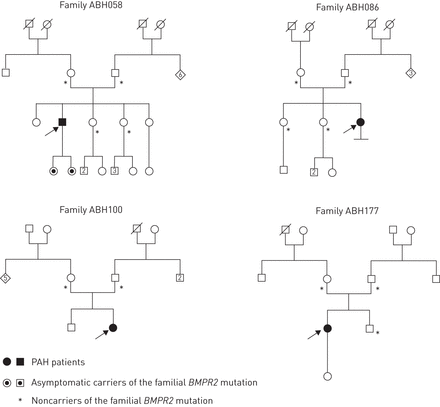
家谱的病人显示新创突变的BMPR2。
十PAH患者家族史的多环芳烃在启动子区域基因突变筛查BMPR2。两个变量意义不明的被确定(在线补充表S1)。这些变异不是人类遗传变异中描述数据库(1000基因组或外显子组变异服务器)和> 400控制等位基因测序显示这些变体。使用生物信息学预测工具MatInspector,我们发现了一个假定的转录因子结合位点受到其中一个变体(c - 1585 g > a)。然而功能分析将有必要描述这些变异的转录活性的影响BMPR2之前我们可以得出他们的潜在的有害影响。
ACVRL1(ALK1),英格在PAH突变发现病人
在诊断、10个病人显示一种零星的多环芳烃和三个病人(来自两个不同的家庭)显示一个家庭形式的多环芳烃提供签名或者家族史的遗传性出血性毛细血管扩张症。ACVRL1(ALK1),英格在这种情况下筛选在第一个实例。的突变ACVRL1(ALK1)被确认在9个10零星的病人和家庭形式的多环芳烃(图1和2)。正如之前报道的(16),的突变ACVRL1(ALK1)通常是局部的外显子10,尤其是在该地区为南盒ALK1蛋白质编码。剩下的零星的病人进行英格突变(在线补充表S1) (21]。这种突变之前描述的遗传性出血性毛细血管扩张症患者(突变报道遗传性出血性毛细血管扩张症突变数据库:http://www.hhtmutation.org)。
此外,84 35岁以下的零星的PAH患者和10人家庭没有突变BMPR2基因被发现的突变筛查ACVRL1(ALK1)。没有发现突变。然而,不明意义的三个变体ACVRL1(ALK1)发现:c.1049-5C > G;c.293A > G;c.314-16T > C。c的变体。29日3一个>G was previously described in HHT patients (variant described in the HHT Mutation Database:www.hhtmutation.org)。
SMAD9,CAV-1和KCNK3PAH患者的突变筛查
十无关的家族PAH患者没有突变BMPR2,ACVRL1(ALK1),英格确定是筛查突变SMAD9,CAV-1和KCNK3。没有发现突变SMAD9和CAV-1(图1和2)。然而,突变KCNK3被确定在两个不相关的患者(图1 b和2)[25]。
作为科学研究,以估计的频率KCNK3零星的PAH突变人口,14额外零星的PAH患者测序KCNK3突变和没有确定突变(25]。
EIF2AK4PAH患者的突变筛查
剩下的八个遗传多环芳烃的家庭,没有突变BMPR2,ACVRL1(ALK1),英格,SMAD9,CAV-1和KCNK3被识别,筛查突变EIF2AK4。没有发现突变(图2)。
遗传咨询和检测PVOD病人
遗传咨询和检测提供100 PVOD患者,其中96人超过18岁在PAH诊断12个家庭的我们发现了19个病人显示PVOD家族的形式。在所有的家庭,我们确定bi-allelic突变EIF2AK4(图2和在线补充表S2)。我们还测试了81例显示零星PVOD。Bi-allelicEIF2AK4突变被确定在7个零星PVOD病人(9%)。
17EIF2AK4突变携带者以前描述(29日),我们报道小说在两个遗传突变确定PVOD病人(c。2609 c > T, p。一个la870Val and c.4910C>T, p.Leu1637Pro mutations; c.3633dup, p.His1212Thrfs*8 and 4318C>T, p.Gln1440* mutations).
所有零星PVOD病例EIF2AK4突变筛选了BMPR2的基因突变,并没有发现突变。最近发现之前的参与EIF2AK4突变PVOD,所有家族PVOD病例筛查BMPR2和ACVRL1(ALK1)突变,和五个PVOD家庭进行筛检KCNK3,CAV-1和SMAD9突变。没有家族中确定基因突变在这些PAH-predisposing PVOD病人。
的一个变种BMPR2被发现在一个PVOD bi-allelic突变患者EIF2AK4。这变种是局部的外显子5 (c.604A > T;p.Asn202Tyr)和不确定在其他多环芳烃或PVOD病人,因此可能代表一个中立的变体。
PAH基因检测的结果和PVOD患者
在PAH-predisposing基因突变的存在
在存在突变PAH-predisposing基因(无论家族病史的病人),病人分为遗传疾病分类肺动脉高压的1.2(集团)(1,2]。多环芳烃因BMPR2,ACVRL1(ALK1),英格,KCNK3或CAV-1突变是一个不完全外显率的常染色体显性遗传疾病。在BMPR2突变携带者,外显率估计为14%,男性为42%,女性(6]。这些患者被告知可能的风险在他们的家庭(父母,兄弟姐妹和后代)发展中多环芳烃。我们鼓励病人与亲人分享这种基因信息,通知他们关于遗传咨询和pre-symptomatic诊断的可能性。
PAH-causing突变的患者有50%的风险传递家族突变后代。在法国,这些患者的生殖选项保持无子女,收养,有孩子没有基因检测(生殖机会),配子捐赠或接受胚胎植入前诊断。目前,男性PAH患者携带BMPR2突变提供胚胎植入前诊断的可能性。由于多环芳烃的危及生命的怀孕的风险,避免怀孕妇女生育潜力的建议。
在没有检测到突变的病人显示零星的多环芳烃
在这种情况下,我们没有告知病人诱发突变已被确定在知识的当前状态。Pre-symptomatic基因测试不能提供给家庭的其他成员。因此,我们建议没有系统筛查其他家庭成员。病人告知它仍可能nonidentifiable突变将在家庭和家庭成员被要求告知我们如果症状暗示多环芳烃发生。
没有突变的病人显示一种家族的多环芳烃
在这种情况下,PAH患者告知一个未知的基因原因可能将在家里,但是,我们无法识别它。因此不可能提出pre-symptomatic诊断和对家庭成员进行胚胎植入前诊断。每个家庭成员都应该意识到他们是潜在的发展中多环芳烃的风险。仔细分析的遗传树允许识别的航空公司的常染色体显性遗传特点仔细随访可以提出。
具体问题的遗传咨询PVOD病人
PVOD由于EIF2AK4bi-allelic突变是一种常染色体隐性疾病待定外显率。因此,患者必须了解PVOD为兄弟姐妹的风险。到目前为止,没有数据的临床表型的杂合子携带者EIF2AK4突变,但是我们现在还没有观察到任何迹象表明PVOD 29中杂合子携带者中确定我们的队列。杂合的一般人群的频率已经估计< 1/8000 (29日]。我们对法律义务告知病人与兄弟姐妹分享这种基因信息,并通知他们关于遗传咨询和pre-symptomatic诊断的可能性。
遗传咨询在高风险的多环芳烃或PVOD病人的亲属
我们的战略使我们能够识别患者遗传条件,因此亲人患多环芳烃或PVOD发展。当在诱发基因突变中确定一个病人,遗传咨询和pre-symptomatic基因诊断可用于亲戚18岁以上。亲戚告知他们携带的风险家族突变,患这种疾病的风险和突变的传播他们的后代。他们也收到完整的信息多环芳烃或PVOD症状,疾病特点和预后。
所有的亲戚都有机会有足够的时间来考虑是否要进行基因测试。此外,遗传的结果可用血液样本后1个月。这个延迟允许亲属的机会改变自己的思想和暂停pre-symptomatic诊断暂时或永久。在法国,pre-symptomatic基因检测多环芳烃和PVOD不是给18岁以下的无症状的亲戚,因为它认为没有预防性治疗多环芳烃或PVOD (表1)。
2014年12月,272 PAH患者的亲属同意pre-symptomatic基因诊断(一级亲属n = 216;二级亲属n = 56)。这使我们能够识别99(36.3%)无症状携带者PAH-predisposing基因的突变:96家运营商BMPR2突变的一个载体ACVRL1(ALK1)突变和两个航空公司的KCNK3突变(图1)。
对于常染色体隐性遗传的传播PVOD pre-symptomatic诊断只是建议携带bi-allelic PVOD病人的兄弟姐妹EIF2AK4突变。基因检测提供了两个兄弟姐妹和他们携带bi-allelicEIF2AK4突变。
我们进行了一项调查在48无症状的亲属为了记录他们的理由接受pre-symptomatic基因诊断:42%进行基因测试通知自己的遗传风险,告知他们的后代的遗传风险35%,10%为目的的生育与可能的胚胎植入前诊断、10%通知家人对他们的基因状态,8%为了知道家庭分支PAH的危险,6%把怀疑的航母航母,6%为了受益于专业化后续如果他们家族突变携带者的6%,帮助研究领域的多环芳烃。一个相对收到遗传咨询但拒绝后续基因测试的心理影响一个积极或消极的结果。然而,17个亲戚(4BMPR2突变携带者)进行基因检测遗传咨询接收他们的结果就再也没有回来。
遗传咨询后亲戚发展中多环芳烃
四个无症状的亲戚(一个男人和三个女人)开发了多环芳烃与延迟24遗传咨询后,41岁的48和96个月。诊断的多环芳烃,三个亲戚有严重的多环芳烃和在纽约心脏协会(NYHA)功能类三世,严重血流动力学障碍(肺动脉平均34 - 70毫米汞柱,心脏指数(CI) 1.66 - -2.5 L·分钟−1·米−2)。一个亲戚被诊断出患有轻微的多环芳烃在2个月后交付(NYHA功能II级、肺动脉平均25 mmHg,可信区间3.77 L·分钟−1·米−2)。所有四个病人正在接受特定的PAH治疗和还活着。
后果的基因导致PAH-predisposing无症状携带者的突变基因
如果家族突变不是在一个相对确定,这个人有相同的发展中多环芳烃的风险。这个人不需要临床监测PAH的风险,也没有将家族突变给他/她的后代。相反,如果家族BMPR2突变是确定在一个相对无症状,这人估计有14%的风险发展的多环芳烃如果42%男性或女性。的外显率ACVRL1(ALK1),英格,KCNK3和CAV-1PAH突变是不清楚。到目前为止,它不可能区分无症状的受试者携带突变将开发多环芳烃从那些永远不会发展疾病。此外,这个人有50%的风险传播疾病导致突变后代。这个人的生殖选项保持无子女,收养,有孩子没有基因检测(生殖机会),有一个配子捐赠或接受胚胎植入前诊断。
后续的无症状BMPR2突变携带者
尽管新的治疗方法的发展,多环芳烃仍然是一个疾病预后不良,无法治愈是可能的,除了在选定的情况下肺移植。然而,最近的研究表明,早期诊断与更好的长期成果(35]。因此,似乎多环芳烃的重要建立早期诊断,尤其是高危人群如无症状携带者PAH-predisposing的突变基因。没有统一的共识或指导如何遵循这个人口。然而,考虑到这些学科发展中多环芳烃的高风险(外显率从14%到42%不等与15到25 /百万在一般人群),和有限的知识特征的人群,似乎有必要为这些人提供前瞻性随访。目前,无症状的个体阳性PAH-causing突变,以及遗传PAH患者的一级亲属谁没有因果变异已确定,提供年度检查超声心动图或出现症状时36]。应该澄清等问题进行纵向研究最优筛选策略和预测多环芳烃在无症状的进展BMPR2运营商(DELPHI-2研究;www.clinicaltrials.gov:NCT01600898)。
最后,我们报告了第一次胚胎植入前诊断在几个广泛引起的多环芳烃的家族病史BMPR2突变(37]。自那以后,胚胎植入前诊断已提供两对夫妇和一个额外的三对夫妇已经申请了这个过程。这种策略可以提供BMPR2突变携带者为了避免基因传输的后代。目前,胚胎植入前诊断不是提出当女人有家族性突变,因为疾病的发生的风险的情况下怀孕,我们缺乏的知识女性的卵巢刺激的影响BMPR2突变。
讨论
在12年的遗传咨询活动中,我们提供基因检测629例显示多环芳烃或PVOD之后在法国转诊中心肺动脉高压。这个系统的研究使我们能够识别192个病人携带PAH / PVOD-predisposing基因的突变。26 PVOD患者被发现携带bi-allelicEIF2AK4突变。此外,我们确认12 PAH突变患者局部ACVRL1(ALK1),两个突变的患者KCNK3来自127个不同的家庭和151病人携带的突变BMPR2。这些突变传遍BMPR2除了外显子13。突变的第一部分中主要确定蛋白(细胞外域和激酶域)只有14%的突变局部细胞质尾域。似乎所有PAH的运营商ACVRL1(ALK1)突变或者个人遗传性出血性毛细血管扩张症的迹象和/或遗传性出血性毛细血管扩张症迹象可以确定他们的家庭成员。这个观察表明,针对质疑遗传性出血性毛细血管扩张症的迹象,应系统地执行所有PAH患者以屏幕ACVRL1(ALK1在这种情况下)在第一个实例。在缺乏个人和家族性遗传性出血性毛细血管扩张症的迹象,筛查ACVRL1(ALK1)似乎是不必要的,除非家庭数据丢失。此外,重要的是要注意,在此期间,我们没有选择屏幕SMAD9,KCNK3和CAV-1的启动子BMPR2患者认为一种特发性的PAH由于家族的低频变异形式的PAH基因和得出结论的有害影响的困难在启动子区域的变异BMPR2。然而,在未来,下一代测序可能允许筛选所有基因参与血管疾病在所有事件中多环芳烃/ PVOD病人。
有趣的是,尽管只有17%的病人显示明显突变携带者特发性多环芳烃与家族PAH患者的89%相比,我们有大致相同的突变携带者绝对数字在这两个组:62突变携带者在特发性肺动脉高压病人和89年的PAH患者家族史。这个观察强调的重要性,提出遗传咨询和检测所有患者多环芳烃被认为是特发性。此外,正如之前报道的Thomson等。(38),我们确定了四个病人携带新创突变BMPR2。因此,设置的新创突变,即使没有患这种疾病的风险上升的一代,后代必须了解他们的风险和应遵循。一些遗传PAH的情况下仍然没有可识别的基因突变。事实上,基因突变在PAH-predisposing并不确定在11%的PAH患者疾病的家族史。这些发现表明,其他(尚未未知)基因可能参与发展的多环芳烃和所有这些剩余的家庭没有发现突变系统地筛选当怀疑新PAH-predisposing基因的报道。
Pre-symptomatic基因诊断提出了所有的亲属突变携带者和272年无症状的亲属PAH基因检测患者咨询,其中99年无症状的亲戚被发现在PAH-predisposing携带突变基因。BMPR2/ACVRL1(ALK1)/英格/KCNK3PAH突变的特点是不完全外显率,到目前为止它是不可能确定无症状突变携带者最终将发展多环芳烃。此外,预防和治疗疾病的治疗和相关遗传或环境因素导致这些突变携带者PAH的发生仍然是未知的。因此,基因检测在家族的亲戚可以告知运输状态突变,但突变携带者将面临其他的恐惧和不确定性,因为我们不能确定他们是否会发展的疾病,发病的年龄或疾病的进展。因此,基因检测的结果在无症状的突变携带者可能有一个重要的心理影响,恐惧和焦虑的感觉。一些亲戚报道的负罪感突变传染给他们的孩子或没有了疾病与其他家庭成员。我们群的研究显示,基因检测的结果也有影响的亲戚不携带家族突变。虽然他们放心没有发展中多环芳烃的风险,并没有将变异后代的风险,一些显示的负罪感,愤怒、懦弱、不公和遗弃。所有这些原因,适当的心理支持是强制性的pre-symptomatic诊断的过程中,必须提供和谨慎咨询关于知道他们的优点和缺点的遗传状态。
它已经表明,早期诊断和及时干预多环芳烃可能转化为更好的长期结果35,39]。Pre-symptomatic遗传筛查可以感兴趣的,因为它将导致小心和常规的临床随访无症状突变携带者和促进早期PAH诊断。因此,在法国转诊中心肺动脉高压,我们提供所有的无症状BMPR2突变携带者项目检测疾病的早期阶段。这个临床筛查包括临床检查、心电图和超声心动图或每当疾病症状发生。然而,没有验证策略,我们启动了一个研究项目2012年3月以来(DELPHI-2研究;www.clinicaltrials.gov:NCT01600898)为了定义最优筛选策略和在这些无症状筛查突变携带者的频率。
最后,识别的BMPR2突变在男性无症状的主题或PAH患者也可能导致产前诊断的考虑或胚胎植入前诊断。这两个技术是用来避免个体的诞生的突变可能导致危及生命的和无法治愈的疾病。这些技术被用于其他疾病,如亨廷顿氏舞蹈症或囊性纤维化,但他们仍然受到辩论中多环芳烃的不完全外显率BMPR2突变。由于这种毁灭性疾病的严重程度和堕胎的潜在影响的父母,我们的团队的技术支持胚胎植入前诊断后在选择伴侣的和伦理的讨论。
总之,BMPR2,ACVRL1(ALK1),英格,KCNK3和CAV-1突变使多环芳烃开发,bi-allelicEIF2AK4PVOD突变引起。我们12年的经验表明,遗传咨询可以实现在转诊中心肺动脉高压。需要更多的数据来验证多环芳烃检测算法在无症状BMPR2突变携带者。
脚注
社论评论看欧元和J2016;47:388 - 389 . DOI:10.1183/13993003.01858 -2015]。
可以从本文的补充材料www.qdcxjkg.com
利益冲突:披露可以找到与本文的在线版本www.qdcxjkg.com
支持支持声明:l . Godinas欧洲呼吸学会(格兰特ltrf - 2013 - 1592)。188bet官网地址y田村(RESPIRE2 - 2013 - 4919)是收件人的人/欧盟RESPIRE2居里夫人研究奖学金。
- 收到了2015年5月7日。
- 接受2015年10月14日。
- 版权©2016人队











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}