





文摘
通过衰减t细胞激活,免疫检查点(ICs)限制最优抗肿瘤反应,IC抑制(ICI)已经成为一个新的治疗各种癌症。t细胞反应是不可或缺的结核病免疫。然而,提高癌症患者的t细胞免疫阻断细胞程序性死亡1 /细胞程序性死亡配体1 (PD-1 / PD-L1)轴可以触发再活跃的潜在的结核病。这种现象似乎与普遍认为提高t细胞免疫结核分枝杆菌将改善该病原体的免疫控制。数据支持这个传闻的人类,但一些小鼠的研究表明,PD-1不足导致严重结核病快速死亡。这些观察保证认真重新考虑什么是有效的结核病免疫和ICs如何为它作出贡献。通过抑制t细胞反应,ICs至关重要,防止过度的组织损伤和维护一系列效应函数。支持这种想法,抑制性受体限制病理学在流感等呼吸道感染,在损失负免疫调节导致进步的免疫病理反应。在本文中,我们分析了ICs的一般机制及其作用,尤其是结核病。我们总结反思的新兴模式和未来研究的方向。
文摘
采用免疫抑制检查站host-directed治疗癌症,但许多报告表明,它会导致再活跃的潜在的结核病。这个观察权证的重新评价保护性结核病免疫和司机再活跃。https://bit.ly/3vi2xu0
介绍
许多host-directed疗法(HDTs)近年来被许可用于治疗传染病和非传染性疾病通过调制的宿主的免疫反应。或许其中最成功的是使用免疫抑制检查站(ICI)在许多癌症的治疗1]。集成电路由一个家族的受体免疫细胞表面表达,尤其是CD3的t细胞,并通过多种机制减弱细胞激活(2]。这些分子是必不可少的在促进外围宽容和防止过度免疫反应,可能导致免疫病理(3]。然而,ICs也能阻碍有效免疫力,如某些抗肿瘤反应的情况下,和他们的抑制已经被证明是一个强大的治疗工具(4]。常用的抑制剂对多种肿瘤类型包括治疗性单克隆抗体,如pembrolizumab和ipilimumab,针对IC通路的程序性细胞死亡1 (PD-1)和细胞毒性T-lymphocyte-associated蛋白4 (CTLA-4),分别。阻止这些通路及抗肿瘤的t细胞,然后能够有效地针对恶性肿瘤细胞,在许多情况下根除肿瘤(5]。
尽管一般来说有利的结果,越来越多的临床报道出现再活跃的潜伏性结核病(TB)的病人接受ICI疗法治疗癌症。多个实验研究在人类和动物系统添加了支持这些观点,和筛查潜在的结核病现在被视为一个重要的预防措施对那些病人接受ICI (6- - - - - -8]。一方面,进步的结核病的发展的背景下,增强t细胞活动有点违反直觉的,考虑到绝对要求人类结核病免疫t细胞(9]。另一方面,这一事实改变通过这里可以直接影响结核病免疫t细胞免疫,虽然消极,让人不得不怀疑同样的途径可以校准产生的积极作用已被证明领域的癌症。还应该注意从一开始,然而,ICs是表达的各种细胞,因此再活跃在这里可能不完全是由于对t细胞的影响。在这里,我们审查的角色IC通路在结核病和抑制的影响,评估结核疾病进展的可能机制引起的这里,和评估的前景改建ICI提高结核病的结果。
保护结核病免疫障碍
保护性免疫力结核分枝杆菌人类是一个复杂的主机和致病因素之间的平衡,还没有完全理解的复杂10- - - - - -13]。事实上,尽管经过多年的研究,相关的防护结核病免疫在很大程度上仍未知(14]。CD4 t细胞所扮演的角色,然而,被广泛接受的关键,受到广泛的数据从动物模型和在艾滋病毒感染的CD4 t细胞耗竭严重削弱了结核病免疫(15- - - - - -17]。同样,肿瘤坏死因子(TNF) -α和白介素(IL) -12 /干扰素(IFN) -γ轴被认为是至关重要的组件,在这些信号通路基因缺陷一直是与疾病进展的风险增加相关18- - - - - -21]。此外,TNF-α阻断剂,用于治疗慢性炎症性疾病,如风湿性关节炎和克罗恩氏病,导致大量结核病例再活跃(22,23]。然而,免疫信号受损导致疾病易感性并不意味着过度将保护(24]。
对大多数人来说,自然免疫力结核病似乎是非常有效的,据估计,只有10%的受感染的个人一生中患活动性结核病病(25]。尽管如此,很大一部分被感染的个体可能会潜伏性感染,表明免疫是不足以防止建立持续感染肺部(26]。一小部分个体,通常与某种形式的免疫功能低如新生儿和艾滋病患者,不能防止感染的早期传播和发展的疾病,称为原发性结核或进步主要结核病(27]。这不同于中等结核病主要影响成人学习,后发生系统性免疫和标记的一代肺腔,方便传输到一个新主机(28]。免疫失败与未解决的发展为活动性结核病的感染和/或在某些个体可能是由于免疫抑制的机制结核分枝杆菌适应性免疫的第一延迟启动,随后逃避的识别结核分枝杆菌来华的细胞,t细胞(29日- - - - - -33]。在问题的根源,结核分枝杆菌展品特殊抗巨噬细胞杀死,肺部免疫防御的第一行(34]。吞噬溶酶体融合在巨噬细胞内,破坏和细胞凋亡结核分枝杆菌防止有效的早期细菌清除率。在人类中,适应性免疫检测感染4 - 5周后,提供充足的时间延长细菌复制(35]。因此,速率辅助(Th) 1型(Th1)淋巴结细胞被激活,迁移到肺被建议作为一种有效的免疫控制的关键因素(36,37]。另一方面,抗原t细胞过继转移到天真的主机之前不加速细菌感染控制,只授予保护感染后7天(38]。此外,没有直接证据表明存在Th1 t细胞在循环或肺保护结核病免疫(39]。
许多机制提出了解释t细胞免疫力的极限结核分枝杆菌。这些包括抑制细胞群的涌入到肺部或持续的抗原刺激,造成细菌高负担,妥协(t细胞功能40,41]。此外,结核分枝杆菌已被证明会破坏抗原表示,部分通过瞄准主要组织相容性复合体(MHC) II限制CD4 t细胞激活(26,32]。这些,连同其他t细胞损伤的可能机制在结核病,都进行了广泛的审查(26,31日,32]。此外,现在认为存在一个多样性的感染个体和群体的层面上,而刚性分类不足以描述肺结核感染的各种表现的特点(42]。记住这些挑战,检查ICs的规定在结核病感染宿主-病原体相互作用可能会提供新的见解,持续感染和活动性疾病的发展。
ICI后再活跃的结核病
ICI治疗癌症的使用代表了主要HDTs概念发展的突破,尽管是非常有效的只有少数患者(43]。此外,一些团体已经报道的发展活动性结核病患者的副作用ICI (图1)[44,45]。第一个报告,由Leeet al。(46),描述再活跃在病人治疗霍奇金淋巴瘤pembrolizumab PD-1抑制剂。接下来,Fujita等。(47]报道的急性结核病治疗后与另一个PD-1抑制剂,nivolumab,肺癌四期。此后许多报告描述类似的观测结果ICI已经出版包括伴随免疫反应的观察。分析周边t细胞反应之前和之后anti-PD-1治疗癌症病人在一个开发结核病再活跃了B乔木et al。(48]。在这里,抗原Th1反应结核分枝杆菌,但不是Th17或CD8 t细胞反应,发现pembrolizumab治疗后3个月。尽管存在结核分枝杆菌特殊抗体,符合感染之前,这些Th1反应ICI之前缺席。虽然只有一个个体的研究,数据的B乔木et al。(48)可能表明提高Th1 t细胞功能活跃,可能会导致发展初等后结核病。然而,如上所述,PD-1表达各种免疫细胞的子集,没有报告。引人注目的是,所有出版物、酒吧,描述结核病的发生由于PD-1 /细胞程序性死亡配体1 (PD-L1)抑制。除了涉及病人最初收到anti-CTLA-4之前继续anti-PD-1,因此不能排除CTLA-4[的参与49]。除了结核,Fujitaet al。(50最近报道了3例鸟型分支杆菌再活跃在肺癌患者接受anti-PD-1疗法,建议再活跃机制在不同的分枝杆菌物种可能是守恒的。最近,三重癌症治疗,包括化疗和免疫治疗同时,与几位结核病例再活跃有关51,52]。美国食品和药物管理局的分析不良事件报告系统在2015年和2020年之间显示72例结核病和非典型分枝杆菌感染13例由于使用PD-1 / PD-L1抑制剂(53]。结合动物实验中,这些数据识别anti-PD-1有害的结核病免疫治疗,有利于疾病再活跃。相比之下,没有流行病学数据表明,CTLA-4抑制引发结核病再活跃(53]。
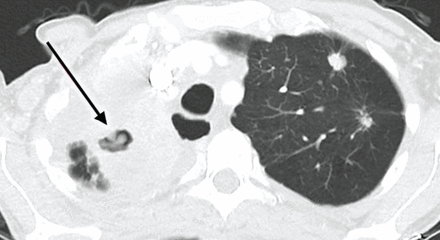
电脑断层扫描显示进步的肺结核病(箭头)的癌症患者的肺nivolumab对待。从[复制45经允许)。
ICs的机制
免疫系统已经进化到抵御感染,然后迅速回到组织内稳态(54,55]。不成比例的免疫反应会造成组织损伤,因此免疫反应的密切监管是必需的。IC分子现在被认为是这个过程的关键部分,通过扮演刹车,避免过度的t细胞活化和随后的免疫病理或自身免疫56]。此外,限制t细胞活动可能保持t细胞克隆为未来的病原体,通过防止activation-induced细胞死亡(3]。IC表达式和t细胞功能障碍之间的关系,然而,复杂。一般来说,任何单一的表达IC分子被认为是t细胞活化的标志而不是疲惫(57,58]。事实上,幼稚t细胞不表达IC分子和他们的感应是直接与t细胞受体(TCR)信号强度59- - - - - -61年]。此外,tissue-resident记忆t细胞,这是高度功能和免疫屏障地点至关重要,通常表达高水平的PD-1 [62年,63年]。疲惫,另一方面,通常定义为有缺陷的效应函数,通常是与IC的持续表达的分子,和几个IC的coexpression分子是指示性损伤的严重性3,64年]。此外,充足的证据表明ICI t细胞活动的后果不是通用的,是依赖于特定通路抑制(3]。换句话说,某些IC通路的封锁,或组合的途径,可能导致不同的t细胞扩张和活动模式。在这里,我们提出一个简短的概述知名IC分子和管理功能的分子和细胞机制(图2)。
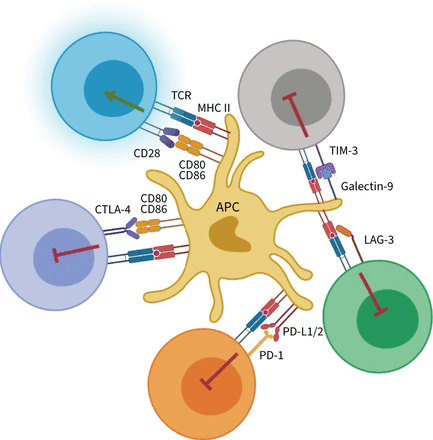
免疫检查点(IC)通路抑制t细胞激活。调节t细胞的反应依赖于与抗原呈递细胞(apc)。同源抗原的表达主要组织相容性复合体(MHC)分子是认可的t细胞受体(TCR)。其次,CD80 / CD86装甲运兵车提供“信号2”在t细胞CD28。在一起,这两个信号诱导t细胞激活(绿色箭头)。相比之下,ICs抑制t细胞激活(红色“阻塞”符号)作为主机策略来防止过度免疫反应或病理的函数以抑制免疫力。CTLA-4:细胞毒性T-lymphocyte-associated蛋白4;LAG-3:淋巴细胞激活基因3;PD-1:程序性细胞死亡1;PD-L1/2:细胞程序性死亡配体1/2; TIM-3: T-cell immunoglobulin 3.
PD-1
PD-1扮演主要角色在维护中央和周边的宽容,和限制t细胞反应(65年]。PD-1发挥其功能通过限制信号通过识别和co-stimulatory CD28分子,它提供了所需的“第二信号”t细胞激活通过绑定的配体CD80和CD86抗原呈递细胞(APC)。由其配体参与PD-1 PD-L1激活酪氨酸磷酸酶SHP2 PD-L2结果,进而抑制信号通过识别和CD28。已经提出的平衡co-stimulatory分子的激活和抑制受体功能作为一个变阻器来调整t细胞反应(66年]。以这种方式阈值t细胞抗原反应的调节(67年]。由nonhaematopoietic细胞表达PD-1配体以及造血的细胞,诱导炎性细胞因子,有助于保持体内平衡控制和防止组织损伤(68年]。然而,肿瘤移植PD-L1/2回应T-cell-derived IFN-γ为了逃避免疫监视和维护一般抗炎环境;这种现象被称为“适应性免疫抵抗”(69年]。因此,响应ICI疗法相关的预先存在的抗肿瘤CD8 t细胞表达PD-1因此束缚PD-L1/2表达于肿瘤细胞(70年,71年]。与这种机制一致,anti-PD-1治疗失败的患者肿瘤展览IFN-γ通路中的基因缺陷(72年,73年]。纵向检查四期黑色素瘤患者的外周血PD-1识别+CD8 t细胞的主要目标PD-1抑制,导致明显的扩张IFN-γ-producing CXCR5+PD-1+(子集74年]。与此一致的是,PD-1封锁在慢性淋巴细胞性脉络丛脑膜炎病毒(淋巴细胞脉络丛脑膜炎病毒)来华的老鼠导致CXCR5的扩张+PD-1+CD8 t细胞(75年]。这是进一步证实了在人类和小鼠组织比较CTLA-4与PD-1抑制(76年]。
封锁PD-1 / PD-L1轴在人类和动物模型已经被证明可以提高免疫控制感染,如疟疾、乙型肝炎和艾滋病病毒(77年]。在长期LCMV-infected老鼠,PD-1 / PD-L1封锁,但不是CTLA-4封锁,显著降低病毒载量由于重新振作进展得筋疲力尽的CD8 t细胞(78年]。在相同的研究中,然而,PD-L1淘汰赛(KO)老鼠非常容易感染淋巴细胞脉络丛脑膜炎病毒,免疫病理死亡迅速,突显出一部分由PD-1轴在限制组织损伤。有趣的是,基因丧失PD-1导致CD8 t细胞终末分化的累积效应在LCMV-infected老鼠(79年]。这一发现演示了一个可能的角色PD-1保护t细胞的数量由疲惫。至关重要的是,这项研究表明,疲劳可以发生在缺乏PD-1,证明分子本身没有定义一个疲惫的状态。此外,尽管PD-1表达高水平,更多的CD8 t细胞终末分化减少响应PD-L1封锁,暗示的疲惫超出阈值t细胞功能不能恢复(57]。还讨论什么精确区分疲惫和t细胞终末分化,和讨论更加复杂,使用的术语可以不同80年]。因此,PD-1在免疫调节中的作用是非常微妙和抑制的影响似乎非常依赖上下文。
CTLA-4
CTLA-4也抑制了t细胞活化与CD28争夺其配体CD80和CD86表达在装甲运兵车81年]。结构相似性CD28和更强的亲和力CD80和CD86允许CTLA-4战胜这些配体,所以限制t细胞CD28激活(81年,82年]。有趣的是,最近的数据表明,PD-1-induced SHP2主要目标CD28、指示功能重叠CTLA-4和PD-13]。为主,早期CTLA-4调节t细胞在淋巴器官和控制激活启动外围组织。老鼠基因KO或antibody-meditated抑制CTLA-4导致异常扩张的几组CD4 t细胞效应,表明CTLA-4的关键作用在调节t细胞扩张和分化83年]。CTLA-4至关重要的功能调控t细胞亚群)和CTLA-4封锁会削弱这一活动(84年]。在亚群,CTLA-4行为通过竞争效应t细胞co-stimulatory配体和通过限制这些分子,消耗他们的可用性在细胞表面通过transendocytosis [85年,86年]。的确,CTLA-4表情亚群的维护需要宽容,那么严重的免疫失调与Treg障碍在人类CTLA-A不足(87年- - - - - -91年]。删除老鼠CTLA-4导致严重的自身免疫性疾病和淋巴组织的发展障碍(92年- - - - - -94年]。这是符合CTLA-4的角色在消除autoreactive t细胞在淋巴结(幼稚t细胞激活95年]。相比之下,PD-1和t细胞免疫球蛋白3 (TIM-3)控制t细胞活化后阶段,这或许可以解释缺乏自身免疫观察小鼠缺乏这些抑制性受体。CTLA-4耗竭小鼠增强antitumoral CD8 t细胞活性和抑制肿瘤微环境内的亚群96年- - - - - -99年]。在人类和老鼠,anti-CTLA-4治疗导致这个理事会的增加+T-bet+Th1-like CD4细胞的表型效应t细胞以及疲惫CD8 t细胞(76年,One hundred.,101年]。矛盾的数据存在,然而,关于CTLA-4封锁在感染控制的影响。CTLA-4抑制不增加阻力刚地弓形虫而小鼠疟疾感染恶化[102年,103年]。相比之下,CTLA-4抑制加速清除单核细胞增多性李斯特氏菌在老鼠和增强HIV抗体诱导在猴子104年,105年]。尽管anti-CTLA-4和anti-PD-1导致CD8 t细胞的扩张,独自anti-CTLA-4似乎扩大CD4 t细胞室,突显出对比CTLA-4 t细胞扩张的模式与PD-1封锁[76年]。
LAG-3和TIM-3
除了PD-1和CTLA-4,其他分子已成为潜在的ICI的目标,包括淋巴细胞激活基因3 (LAG-3)和TIM-3 [106年]。LAG-3结构类似于CD4和结合MHC II与亲和力高于CD4分子,可能抑制信号传输通过其胞质域(107年,108年]。此外,t细胞内稳态LAG-3负调控通过Treg-dependent机制(109年]。感染和癌症,coexpression LAG-3和PD-1负调节t细胞反应,这可以通过联合封锁[补救110年- - - - - -113年]。galectin-9 TIM-3吸引它的配体,抑制t细胞功能有选择性地诱导细胞死亡的IFN-γ-producing Th1细胞(114年]。其他TIM-3配体包括磷脂PtdSer表示对凋亡细胞,alarmin高机动性组框1 (HMGB1)与DNA结合释放死亡细胞和醣脂类东航细胞粘附分子1 (CEACAM1)已知高度表达的肿瘤细胞(115年]。LAG-3一样,CD8 t细胞激活是由coexpression PD-1 TIM-3,和功能可以恢复双重封锁[116年]。TIM-3还可以作为一种抑制性受体等固有细胞自然杀伤(NK)细胞,巨噬细胞和树突细胞(dc)。订婚的TIM-3 NK细胞显著降低细胞毒性的能力(117年]。在巨噬细胞,TIM-3受损的toll样受体介导细胞因子的过度生产,而封锁TIM-3增强巨噬细胞的激活,导致严重脓毒症(118年]。TIM-3的差别相一致,对这些患者外周血单核细胞(PBMCs)与脓毒症的严重程度相关,指着TIM-3的保护作用抑制过度炎症(118年]。Anti-TIM-3治疗改善通过促进生产CXCL9 CD103 anti-tumoural响应+dc与肿瘤细胞碎片(接触后119年]。TIM-3抑制的影响可能取决于细胞类型封锁不是细胞选择性。
调制ICs的结核病
表达PD-1 / PD-L1调节在活动性结核病
表达谱的ICs结核病患者的特征广泛多年来,尽管研究主要集中在PD-1 / PD-L1轴。PD-1表达一直是增加循环活动性结核病患者t细胞疾病与健康对照组相比(120年- - - - - -123年]。有趣的是,PD-1及其配体的表达(PD-L1和PD-L2)也显著增加单核细胞在积极感染(121年,124年,125年]。此外,一些研究已经表明PD-1表达式直接与细菌负荷和IFN-γ响应的大小,表明抗原水平可能是驱动因素(126年,127年]。事实上,PD-1的CD4 t细胞产生Th1细胞因子表达增加痰检阳性结核病患者相比,涂阴病人和潜伏性感染对象(127年]。在这里,刺激结核分枝杆菌抗原诱导PD-1表情结核分枝杆菌特殊技能CD4 t细胞,这表明其表达式可能直接调节的抗原。这些观察最近扩展到结核病感染的肺组织,PD-1表达最高在t细胞表达的标记组织实习,CD103和/或CD69 [8]。然而,尽管PD-L1表达普遍存在,免疫组织化学染色发现PD-1表达式是缺席的干酪样肉芽肿,可能表明一个角色在限制免疫病理和肉芽肿PD-1进展(8,49]。
早期研究比较CTLA-4表达阴性结核病患者和健康对照组之间以前接触结核分枝杆菌发现CTLA-4表达式在结核病患者比对照组显著降低(128年]。在同一病人组,CTLA-4表达显著调节白介素刺激或il - 10中和。另一份报告未能发现CTLA-4表情如果PBMCs从活动性结核病患者或患者完成治疗129年]。然而,CTLA-4和IFN-γ表达增加卡介苗(BCG)刺激后,这是更明显的患者在治疗结束与新诊断病例。最近,考试的结核分枝杆菌特殊技能CD4 t细胞的表达显著升高透露CTLA-4和PD-1这些细胞(130年]。增加CTLA-4-expressing亚最近在对象被描述活动性肺结核,这减少了治疗后(131年]。
类似的模式中执行的有限研究其他IC分子。LAG-3表达调节猕猴与活动性疾病的肺部,但不是那些潜伏性感染,在人类发现肺granuloma-associated t细胞(132年]。然而,它并不是在t细胞在血液中检测到,并提出这个问题,IC分子的表达可能在不同的血液室,他们常常以人类的地方,疾病的网站。TIM-3表达更高的CD4和CD8结核病血液中的t细胞为活动性疾病患者与健康对照组相比(133年和与疾病严重程度有关134年),在一项研究中观察到与PD-1 [122年]。综上所述,这些数据支持ICs在调节免疫反应的作用在人类结核病,但宿主免疫力的净效应还不清楚。
集成电路表达式表达下调,以应对结核病治疗
生物标志物的鉴定来确定结核病治疗疗效近年来获得了极大的兴趣(135年,136年]。几项研究已经表明PD-1, CTLA-4在外周血和TIM-3表达结核治疗后明显下降。结核—艾滋病毒群,并发抗逆转录病毒疗法和结核病治疗显著降低PD-1 CTLA-4表达CD4 t细胞抗原(130年]。这是进一步证实数据显示在IFN-γ-expressing PD-1表达式只是减少了结核分枝杆菌特殊技能CD4 t细胞治疗后,不是CD8 t细胞(127年]。PD-1表达治疗期间的下降呈负相关的比率IFN-γil - 4,可能表明恢复保护性免疫属性(125年]。另一项研究表明,效应t细胞表现出最大的减少PD-1表达治疗后,没有观察到的差异的亚群(120年]。重要的是,治疗PD-1轴的先天差别也引发对这些细胞,作为PD-L1和PD-L2差别明显对这些巨噬细胞与成功治疗(121年]。NK细胞,PD-1表达减少治疗后2个月开始(137年]。最后,表达PD-1和TIM-3 CD8 t细胞中表达下调结核分枝杆菌来华的老鼠接受治疗后(138年]。虽然没有得到全面的集成电路表达式的优惠减少抗原细胞表明消除抗原刺激的可能机制,至少对CD4 t细胞。
ICs增强效应的抑制功能
考虑IC分子的抑制特性,提出,他们的抑制增强效应函数在结核病(图3)。在小鼠模型中,CTLA-4封锁导致淋巴结中淋巴细胞数量增加,antigen-induced IFN-γ分泌在体外(139年]。然而,这并不影响BCG和肉芽肿形成的间隙。在体外封锁的CTLA-4扩大从受试者活动性结核患者亚群最近显示增强IFN-γ生产和扩散结核分枝杆菌特殊t细胞,提高巨噬细胞的死亡结核分枝杆菌(131年]。一般来说,在体外抑制PD-1 / PD-L1轴提出可能的改进先天和适应性细胞因子的生产和杀戮的潜力。抑制PD-1及其配体,例如,提高脱粒和IFN-γ生产从结核病患者CD8 t细胞和NK细胞的126年,140年]。这种效应可以通过同时co-stimulation进一步增强CD8 t细胞(126年]。两个最近的报告证实了这些发现结核性胸膜炎患者[收集的样本中125年,141年]。封锁PD-1通路增加IFN-γ-producing t细胞的频率以及CD8 t细胞脱颗粒(141年]。具体来说,PD-L1抑制增强CD8 t细胞对促炎症巨噬细胞细胞毒性与抗炎巨噬细胞(124年]。PD-1阻止细胞凋亡的抑制作用结核分枝杆菌特殊技能IFN-γ-producing t细胞取自结核病患者(125年]。因此,虽然PD-1表达式可能保持总体t细胞功能,抑制PD-1可能恢复效应函数在精疲力竭的人群(由于持续的抗原刺激)出现在结核病患者。
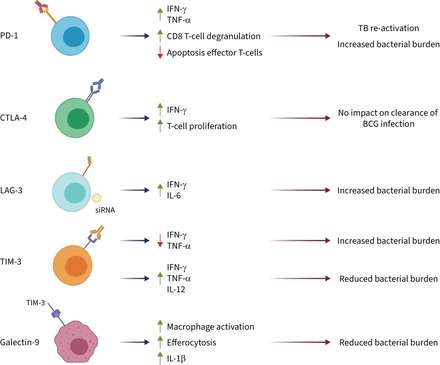
影响免疫抑制检查站(ICI)肺结核(TB)。抑制IC对结核病病理分子和下游的影响。封锁的抗体细胞程序性死亡1 (PD-1),细胞毒性T-lymphocyte-associated蛋白4 (CTLA-4)和t细胞免疫球蛋白3 (TIM-3)。淋巴细胞活化基因3 (LAG-3)是被小干扰RNA (siRNA)。Galectin-9显示其配体TIM-3迷人。干扰素:干扰素;肿瘤坏死因子:肿瘤坏死因子;IL:白介素;波士顿咨询集团:卡介苗。
沉默的LAG-3 lung-derived CD4 t细胞取自猕猴显著降低结核分枝杆菌负担培养巨噬细胞,同时也促进IFN-γ和il - 6生产(132年]。几项研究也调查了调制TIM-3加强结核病的免疫力,有些相互矛盾的结果。例如,TIM-3封锁或消融在老鼠增强t细胞功能和适度降低细菌的负担,而刺激TIM-3 / galectin-9轴促进巨噬细胞的激活和还限制细菌复制由IL-1β分泌(142年- - - - - -145年]。与这些研究结果相反,另一项研究报道,TIM-3+t细胞从结核病患者更强有力地控制结核分枝杆菌巨噬细胞与TIM-3相比的增长−t细胞(133年,146年]。这里,击倒TIM-3通路使用沉默RNA被发现损害IFN-γ生产而刺激进一步加强。虽然需要更多的工作来解决其中的一些对比观察,他们一起突出IC通路之间的差异并提出潜在的宿主-病原体相互作用在调节不同的角色。
PD-1 / PD-L1信号损失加重疾病
无节制的炎症和肺是极其敏感的抑制信号必须减少组织损伤(147年]。失去PD-1轴一致导致结核疾病的恶化。PD-1-deficient老鼠非常容易结核分枝杆菌感染和死亡迅速与野生型小鼠相比143年,148年]。严重坏死性肺炎开发这些PD-1 KO小鼠的肺的特点是大量嗜中性粒细胞浸润和更高水平的il - 6, IL-17和IFN-γ148年]。在另一项研究中,结核分枝杆菌PD-1-deficient感染小鼠与减少巨噬细胞自噬,增殖抗原的淋巴细胞受损和Treg活动增加149年]。随后,使用过继转移和损耗,这是表明减少生存被发现由PD-1缺乏CD4 t细胞CD8 t细胞与未成年人的角色,因为损耗的CD4 t细胞获救PD-1 KO小鼠早期死亡率和严重的肺部病理学(6]。同样,在猕猴加剧PD-1抑制结核病更大尺寸的肺部肉芽肿(150年]。然而,在此系统中,它是伴随着频率的增加结核分枝杆菌IFN-γ特殊CD8 t细胞表现出增加产量和2 anti-PD-1-treated肉芽肿的动物。结核分枝杆菌特殊CD4 t细胞出现的影响,作者得出结论,PD-1封锁加剧疾病主要通过CD8 t细胞。相比结核分枝杆菌PD-1-deficient老鼠控制卡介苗感染更有效地比野生型小鼠(151年]。这表明,在面对一个减毒株,PD-1表达式可能限制间隙,但致命的毒株结核分枝杆菌需要适度的炎症。然而,如前所述,anti-PD-1治疗人类与非典型分枝杆菌感染的风险增加有关,通常被认为是毒性低于结核分枝杆菌(152年]。随着明显差异的作用机制PD-1封锁在猴子和老鼠模型中,这些发现谨慎的直接翻译研究在不同模型和人类结核病。
增加的IFN-γ未能提供保护在活的有机体内或在体外和证据表明IFN-γ可能损害保护性反应,这一切似乎与IFN-γ被不可或缺的结核病免疫的概念(153年- - - - - -155年]。与此一致的是,过度的IFN-γCD4 t细胞已被证明加速死亡在老鼠身上,可以预防的命运PD-1表达式(7]。使用一个在体外肉芽肿模型,PD-1封锁TNF-α产量显著增加结核分枝杆菌来华的人类PBMCs [8]。这导致了细菌生长,可以减轻TNF-α中和。潜在的过度TNF-α可能推动巨噬细胞功能丧失,TNF-α是为了诱导坏死结核分枝杆菌来华的巨噬细胞由活性氧的生产(156年,157年]。然而,在这些实验TNF-α的来源并不成立。最近,PD-1缺乏与孩子与结核病有关低IFN-γ反应(158年]。此外,可能还有其他的有害后果PD-1缺陷,如PD-1信号的要求的亚群在扩张结核分枝杆菌感染(159年,160年]。尽管亚被认为抑制保护性免疫力严重萎缩的人口可能影响CD4 t细胞启动和激活(161年]。因此,尽管PD-1信号在结核病免疫反应,似乎是重要的机制可能是复杂的和上下文特定的。
PD-1-expressing CD4 t细胞与保护性反应
越来越多的数据显示集成电路表达式,包括PD-1,并不仅仅是一个标记的疲惫,而是有必要维持结核的t细胞功能。一个具有里程碑意义的研究雅佳et al。(162年显示的两倍结核分枝杆菌特殊CD4 t细胞在小鼠的肺被保留血管与实质。CD4 t细胞的实质表现出更高水平的PD-1表达,而在脉管系统主要表达了杀手细胞凝集素受体G1 (KLRG1)、终端分化的标志(163年]。更高水平的表达的脉管系统子集T-bet和生产IFN-γ更加智能化。然而,当转移到结核分枝杆菌实质PD-1 T-cell-deficient来华的老鼠嗨KLRG1罗CD4 t细胞迁移到实质,他们比PD-1更有效地限制细菌生长罗KLRG1嗨血管。同样,接种H56亚基授予高级保护后续结核分枝杆菌挑战,与PD-1的感应+KLRG1−CD4 t细胞(164年]。这PD-1+KLRG1−高度增殖的子集)居住的地方到肺,产生更多的- 2和IL-17不如PD-1 IFN-γ和TNF-α−KLGR1+子集,更短的寿命,对过继转移到不能增殖结核分枝杆菌来华的主机(165年]。因此,结核分枝杆菌特殊技能PD-1+比KLRG1 CD4 t细胞带来更多的保护+同行当过继转移(166年]。在一个单独的研究抗原特异性的重要性,多个的注入结核分枝杆菌早期抗原分泌抗原目标6 kDa (ESAT-6)或Ag85B导致upregulation PD-1 CD4 t细胞特定的抗原,但只有Ag85B导致肺部细菌的减少负担(167年]。这些数据,显示功能和保护t细胞可以表达PD-1,表明它并不是严格疲惫TB-specific t细胞上的一个标志,虽然机械的细节尚不清楚。相比之下,疲惫的CD8 t细胞表达高水平的PD-1 TIM-3 LAG-3 IFN-γ较少和2,和与受损的相关控制结核分枝杆菌复制(168年]。CD4 t细胞的存在是必要的,以防止疲劳和促进效应CD8 t细胞,导致增强的承认结核分枝杆菌来华的巨噬细胞。在猕猴透露详细调查结核分枝杆菌特殊技能CD4 t细胞表达高水平的PD-1和CTLA-4 bacilli-containing核心内的肉芽肿,但未能穿透(169年]。最近,同一组发现PD-1封锁并不影响CD4 t细胞渗透肉芽肿的中心,这表明PD-1表达式不影响t细胞贩运到这个区域(150年]。这些发现强调定位的重要性的保护性反应感染部位的镜子并可能缺乏PD-1表达周围坏死肉芽肿中观察到人类。
ICI在结核病的影响
ICI重新激活结核病的观察对疾病病理生理学和提出了有趣的问题的一些基本假设关于保护性免疫。最重要的是:为什么这里有效癌症治疗而导致再活跃的结核病?,这是合理的允许pathogen-specific特征结核分枝杆菌hyperinflammatory环境中茁壮成长(77年),如由ICI。肺的破坏是一个特征明显的标志在活动性肺结核肺结核,由基质金属蛋白酶(MMPs) (170年,171年]。空化被认为是一个关键的过程中生成bacilli-containing气溶胶和促进传播,和结核分枝杆菌可能受到选择压力保持有力的t细胞反应这一过程(172年,173年]。因此,增加Th1反应带来的ICI可能夸大细胞因子释放和MMP的表达,导致组织退化和扰乱微妙的平衡检测的宿主与病原体相互作用存在于肺。这个概念是由观察不同抑制性受体也作为结核病免疫控制的重要调节器。老鼠缺乏趋化因子巨噬D6或- il - 1系统的调节器,人数/ il - 1受体8 (TIR8),迅速被低剂量结核分枝杆菌挑战伴随着相当大的局部和全身炎症(174年,175年]。有趣的是,这两项研究没有显示出差异结核分枝杆菌增长动力学KO和野生型群体之间,表明结核病是由hyperinflammation没有细菌生长。在人类中,经典的流行病学调查表明,强烈的结核菌素反应在童年与风险增加有关结核病在以后的生活中,这意味着疾病协会与提高免疫反应(176年]。因此,这里观察到的效应功能的恢复在体外可能会加剧疾病而不是改善控制(图2)。此外,抑制PD-1和CTLA-4信号与自身免疫异常t细胞活化的结果(3]。Autoreactive t细胞据报道,提高结核病患者和肉芽肿的病理是经常在自身免疫性疾病的观察结节病(177年,178年]。可信的,这里可能在进一步加大autoreactivity和增加组织破坏,尽管事实上CTLA-4封锁似乎并没有因为这可能反对这一假设。
另一方面,一些数据了,尤其是来自动物模型,暗示PD-1表达式实际上可能与增强的阻力结核分枝杆菌。可以想象,结核病延迟是由PD-1维护+t细胞功能抑制。在这个场景中,PD-1封锁导致t细胞功能障碍的特点是hyperinflammatory反应导致崩溃的免疫控制。一个重点突出在这方面是时机。结核病的PD-1实验动物模型在下面进行了突出显示可能是更具有代表性的主要人类结核感染(179年),而ICI-associated结核病在人类几乎肯定是初等后疾病。PD-1-expressing t细胞可能有不同的角色和功能能力在这些不同的疾病,可能其他免疫表达艾多酷子集。事实上anti-PD-1恶化疾病期间管理的早期感染小鼠和猴子,以及潜在的人类,表明PD-1至关重要的监管感染的免疫力在两个阶段。虽然机械的细节不清楚,数据可以相互矛盾的,几个独立的研究表明PD-1表达式与t细胞归巢到肺实质,即。感染的网站。此外,在这些研究中,保护PD-1+t细胞是位于IFN-γ低于他们的PD-1产生的实质−同行;因此,随着免疫反应的质量,这些发现说明其空间组织的重要性。因此,尽管这里在体外似乎恢复经典结核分枝杆菌特殊技能Th1函数(图2),这些功能可能不是合适的,至少在某些情况下。此外,PD-1封锁可能保护性免疫的某些方面产生负面影响。
最后,重要的是要注意,结核病再活跃是一种针对癌症的ICI的副作用,我们所知,这里从来没有结核病治疗主要是用于人类。因此,潜在的癌症治疗中使用的剂量是不适合提高结核病治疗结核病免疫和调整剂量可能会产生有益的影响。记住,ICs更多的作为一个拨开关,影响结核分枝杆菌感染可能在不同的浓度的不同抑制剂(66年,67年]。此外,再活跃引起PD-1封锁发生在没有抗生素。可能PD-1封锁或其他ICI一起可以利用传统的结核病药物治疗来支持潜在或者活动性结核病感染的冲销。更多的抑制剂目前正在调查用于癌症治疗和其效用在结核病仍有待探索106年]。潜在的候选人,实验数据表明,TIM-3-blocking代理商特别是用于结核病可能有一些好处。相反PD-1 KO小鼠,TIM-3缺陷增加生存在结核感染和抑制这种途径减少细菌负担(143年]。实验数据缺乏和许多其他的IC通路,并需要进一步的调查,以确定潜在的目标ICI提高主机控制结核病。
结论
当前的范式,推动Th1反应的增加可能导致更大的控制结核病是挑战的新兴证据anti-PD-1-associated结核病再活跃(图3)。总的来说,针对结核病的观察再活跃anti-TNF-α疗法后,一方面,和anti-PD-1治疗,另一方面,看来一个窗口的保护性免疫存在于人类,坐在两个极端之间的不足和过度的t细胞免疫(180年]。这种最优平衡反应代表强劲包含有效的免疫控制结核分枝杆菌限制重要负监管防止天平倾斜支持hyperinflammation和疾病进展。ICI能否杠杆来调整这种反应还有待观察。
可共享的PDF
脚注
利益冲突:没有宣布。
支持声明:这项工作是由Wellcome战略核心奖(201433 / / 16 /),威康高级研究奖学金(210662 / Z / 18 / Z)和撒哈拉以南非洲结核/艾滋病毒网络研究卓越。资金信息,本文已沉积的Crossref资助者注册表。
- 收到了2021年9月17日。
- 接受2022年4月6日。
- 版权©2022年作者。
这个版本分布在Creative Commons归因执照的条款4.0。
引用
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵











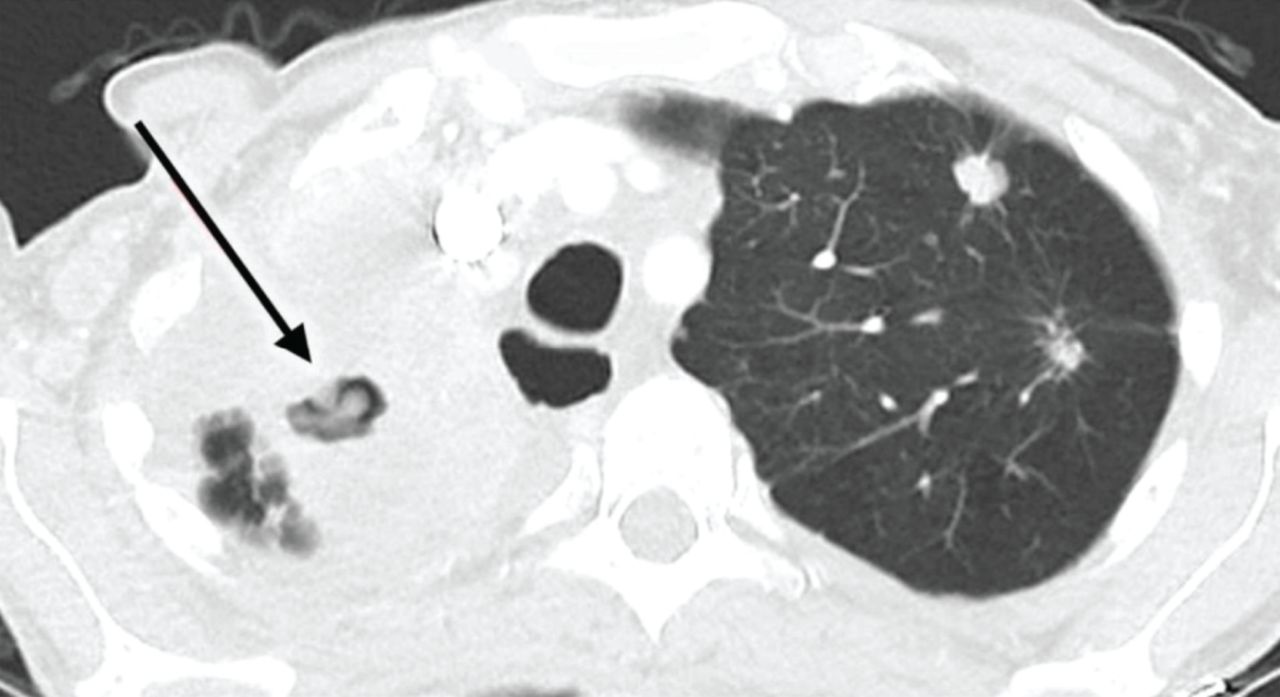
![Computed tomography scan showing progressive tuberculosis disease (arrow) in the right lung of a cancer patient treated with nivolumab. Reproduced from [45] with permission.](http://www.qdcxjkg.com/content/erj/60/5/2102512/F1.large.jpg?width=800&height=600&carousel=1){kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}