





摘要
肺动脉高压(PAH)是一种罕见的呼吸困难-疲劳综合征,由肺血管阻力进行性增加并最终导致右心室(RV)衰竭引起。尽管有广泛的肺血管重塑,PAH患者的肺功能通常保存良好,有过度通气和生理死空间增加,但肺力学变化很小,只有轻度至中度低氧血症和低碳酸血症。低氧血症主要是由心排血量减少引起的混合静脉氧张力低引起的。低碳酸血症主要是由化学敏感性增加引起的。PAH的运动限制是心血管的,而不是呼吸或肌肉的。PAH中肺血管疾病的程度是由多点肺血管压力-流量关系和红细胞压积校正来定义的。PAH中脉动肺血管压力-流量关系可用于评估右心室液压负荷。这种分析既可以在频域,也可以在时域。PAH中的右心室通过增加收缩力来适应后负荷的增加,以保持其与肺循环的耦合。当这一homeometric机制耗尽时,RV膨胀以通过另一种异质计量机制来保持流量输出。 Right heart failure is then diagnosed by imaging of increased right heart dimensions and clinical systemic congestion signs and symptoms. The coupling of the RV to the pulmonary circulation is assessed by the ratio of end-systolic to arterial elastances, but these measurements are difficult. Simplified estimates of RV–pulmonary artery coupling can be obtained by magnetic resonance or echocardiographic imaging of ejection fraction.
摘要
肺动脉高压(PAH)是一种以肺循环、肺力学、气体交换和右心测量为基础诊断的呼吸困难-疲劳综合征,其生理基础被重新解释和更新https://bit.ly/3EhDf24
介绍
肺动脉高压(PAH)是一种罕见的呼吸困难-疲劳综合征,由于进行性增加肺血管阻力(PVR)和最终右心室(RV)衰竭。PAH的诊断依赖于排除引起肺动脉压(PAP)升高的心脏、肺或血栓栓塞原因,然后进行右心导管检查,显示平均肺动脉压(mPAP) >20 mmHg,楔形PAP (PAWP)≤15 mmHg, PVR≥3 Wood单位[1]。在大约一半的病例中,多环芳烃是特发性的[1]。尽管过去几十年治疗方法有所进步,但多环芳烃仍然无法治愈[2]。
多环芳烃的组织病理学特点是小动脉内膜增生,演变为同心或偏心的层状硬化、内侧肥大和外周增生,有不同的炎症反应,偶见纤维蛋白样坏死[3.,4]。晚期PAH可能与所谓的丛状病变有关,丛状病变包括通道增生,内皮细胞和炎症细胞聚集在闭塞的小动脉附近。这些方面在图1.丛状病变散在;倾向于多生动脉,即垂直于大肌肉或弹性肺动脉的分支。它们与PVR的增加没有关系。在大约三分之二的病人身上找不到它们[4]。多达三分之一的PAH患者也可能出现微血栓病变,同样与PVR增加没有明显关系[4]。内侧肥大与急性前列环素诱导的PVR降低相关[5]。
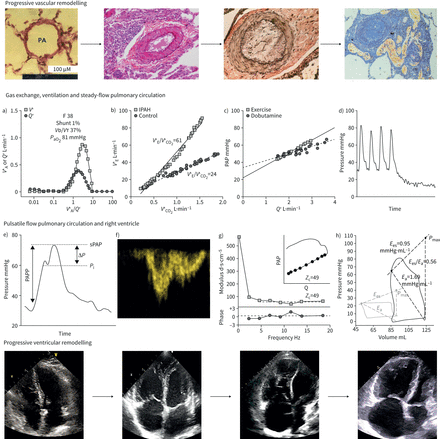
肺动脉高压(PAH)患者肺循环(上一排)和右心(下一排)的进行性重塑,其中包括典型的肺通气和气体交换、稳定血流和脉动血流血流动力学以及右心室(RV)功能测量。显微视图显示血管重构从正常(左)到内侧肥大,三层血管壁增厚和丛状病变(右上)。心脏重构分别由二维四室超声心动图右房和心室扩张以及室间隔移位心室扩张来定义。箭头表示假设的疾病进展,右心室和右心房尺寸不断扩张。介于两者之间的是严重特发性多环芳烃(IPAH)典型患者的测量值:a)肺泡通气准正态分布(V”一个)及灌注(问的函数V' /问的关系,动脉氧分压(PaO2)和生理死空间(死空间体积(VD)/潮气量(VT)),但会增加V'一个;B)增加通风(V”E)作为二氧化碳排放量的函数(V”有限公司2)在运动时;c)普氏调整平均肺动脉压(PAP)与心排血量的函数关系(问’)通过运动或多巴酚丁胺增加,显示斜率下降,外推压力截距增加(n=7例患者);d)单动脉闭塞后PAP衰减曲线显示拐点增大;e)右心室压力曲线随脉压升高、收缩期晚期达到峰值和增强指数升高;f)多普勒右心室流出道血流,加速时间缩短,收缩中期减速;g)肺动脉阻抗谱,或压力/流量和相位随频率的函数关系,随0 Hz和高频阻抗及低频负相角的增大而上升,特征阻抗(Zc)在频域或时域内;h)收缩末期弹性增加的右心室压力-容积循环(E西文)出现动脉弹性增加(E一个),但减少了E西文/E一个与对照组相比增加了尺寸。PAPP:肺动脉脉压;sPAP:收缩压;P我:上行程压力的变化。由Andrew Peacock(英国格拉斯哥区域心肺中心)和Michele D’alto(意大利那不勒斯Monaldi医院)提供组织学视图和超声心动图。图c)转载自[34],经允许。
虽然多环芳烃患者肺循环广泛重构,但这些患者的症状和结局在很大程度上取决于右心室结构和功能的改变[6,7]。多环芳烃作为双侧肺/心脏实体的概念总结在中央插图的上下两排(图1).
本文综述了多环芳烃的两个方面的生理基础,重点是该病症的特发性形式,以避免合并症的混淆效应。
肺力学,通气和气体交换
多环芳烃肺功能测试显示肺容量轻度受限,肺对一氧化碳的扩散能力无或仅轻度下降(DLCO)已记录在经严格确认的IPAH病例登记册内[8]。更重要的减少DLCO在更现代的PAH登记中有报道,其中包括有相关疾病的老年患者、吸烟、临床无症状的心血管或呼吸疾病以及不太严格的血流动力学定义[9,10]。晚期多环芳烃患者小气道阻力的增加可能是动态恶性充气和运动性呼吸困难的原因之一[11,12]。呼吸肌力量亦有下降的报告[13]。然而,肺力学和呼吸肌功能的这些改变似乎并不会限制有氧运动能力[14,15]。
多环芳烃的动脉血气分析的特征是动脉分压正常或降低(PaO2)和二氧化碳(P华2) [8,9,16]。如图2,要么PaO2或P华2约有一半的患者低于正常水平,但严重低碳酸血症比严重低氧血症更常见[16]。低碳酸血症,而非低氧血症,是IPAH生存率下降的独立预测因素[17]。动脉血气,肺活量和DLCO在IPAH和慢性血栓栓塞性肺动脉高压(CTEPH)中同样受到干扰,因此这些测量不能用于区分这两种情况[18]。
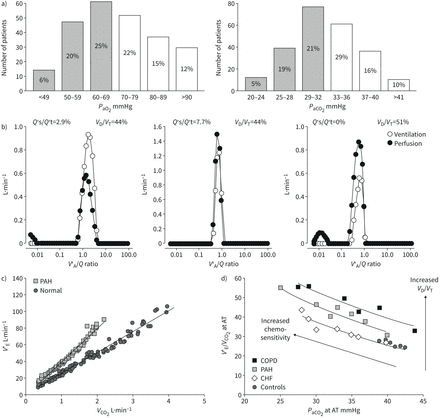
a) 243例特发性肺动脉高压(IPAH)患者动脉血气分布。大多数值都下降了,有51%PaO2<70 mmHg, 45%的P华2<33 mmHg为正常下限(阴影列)。b)肺泡通气(V”一个) /灌注(问’)的分布在IPAH患者的正常化程序之前和之后修正为增加V”一个和减少问,以及中年人的对照(从左到右)。在IPAH患者中,V”一个/问'分布向更高的方向移动V”一个/问”,但V”一个和问模式保持匹配。IPAH患者与健康对照组均有少量至低灌注V”一个/问”。IPAH患者也有少量分流(V”一个/问' =0),随着对高的校正而增加V”一个和低问”。c)通风(V”E)作为二氧化碳(CO .)的函数2)在12例PAH患者和10例对照组中,根据个体间差异调整输出。V”E休息时增加,运动时更明显。d)策划V”E/V”有限公司2作为动脉二氧化碳张力的函数(P华2)在运动无氧阈值(at)时,发现死空间通气增加(死空间容积(VD)/潮气量(VT))和化学敏感性是导致PAH和COPD患者通气增加的原因,或仅在心力衰竭患者中增加化学敏感性。PAH和COPD患者动脉至潮末氧张力梯度增加>5 mmHg(正常上限)。转载自[16]和[25]具有权限。
二十世纪八十年代,人们利用多重惰性气体消除技术探索了多环芳烃中异常血气的生理学[19- - - - - -21]。肺泡通气(V”一个) /灌注(问’)匹配被保留,没有额外的通气或灌注模式比正常更高或更低V”一个/问”。由Bohr-Enghoff方程计算的生理死空间增大。休息时和运动时的低氧血症是由混合静脉氧张力降低(PO2)由于心输出量低,加上灌注分布轻度不均匀[19- - - - - -21]。其中2例患者使用钙通道阻滞剂硝苯地平后灌注增加至低于正常水平V”一个/问’,但这种气体交换的改变与血压的下降无关PaO2,因为心排血量增加和混合静脉PO2[20.]。偶有严重的动脉低氧血症,可通过卵圆孔进行右至左心脏分流术[20.]。肺泡-动脉扩散无限制PO2梯度(19- - - - - -21]。PAH中的动脉低氧血症是“血流动力学低氧血症”的一种形式,即主要由低心排血量的混合性静脉低氧血症引起的动脉低氧血症[16]。
分布的V”一个/问在健康受试者和患有IPAH的患者中图2.的V”一个/问在校正通气增加和心排血量减少后,IPAH患者的分布被重新计算,这是可能使用该方法的数学模型[22]。可见其用意V”一个/问随着两只手都收紧,又恢复了正常V”一个和问'模式,但分流(V”一个/问' = 0)增加。后者的发现可以用两者的增加来解释问’并且减少了V”一个[16]。
PAH患者在休息时、运动时甚至睡眠时过度通气,与增加的生理死空间和化学敏感性有关[16]。对低氧血症和高碳酸血症的通气反应增加的测量已经证明了多环芳烃的化学敏感性增加[23]。由Bohr-Enghoff方程计算的生理死空间对增加的通气很敏感[24]。多环芳烃中增加的化学敏感性和死空间对过度通气的各自贡献可以通过绘制二氧化碳(CO)的通气当量来分类2)(通风(V”E)与有限公司2输出(V”有限公司2))作为潮末或的函数P华2[25) (图2).的上升V”E/V”有限公司2和P华2曲线随动脉至潮末CO的增加而增加2张力梯度超过正常值上限5 mmHg表明生理死空间增加[16]。仅比正常CO略高2多环芳烃中通常存在张力梯度[26]。因此,多环芳烃的过度通气主要是由于增加的化学敏感性。
多环芳烃化学敏感性增加的机制仍未完全了解[27]。休息时和运动时的通气与多环芳烃无关PaO2,P华2或pH值[28],但与右心室舒张刚度密切相关[26]。这一观察结果与PAH患者微神经图研究显示的交感神经系统活性增加一致[29,30.]。都增加了交感神经的激活V”E/V”有限公司2多环芳烃预后较差[30.,31]。
临床意义
PAH中的肺气体交换保存完好。这些患者的换气过度和血气改变主要是由心排血量减少、生理死空间增加和化学敏感性增加共同引起的。
肺循环:稳流血流动力学
肺血管阻力
多环芳烃的定义取决于PVR的测定[1]。然而,由于流量和压力诱导的血管重新招募和/或扩张,PVR随着心排血量的增加而降低[32,33],因此,多环芳烃或左心衰肺动脉高压患者的肺循环阻力特性可以通过多点压力-流量关系更好地定义。这种方法已被用于揭示前列环素治疗多环芳烃的作用[34]。在该研究中,肠外注射epoprostenol降低了map -心排血量关系的斜率,而静息PVR保持不变(图3).
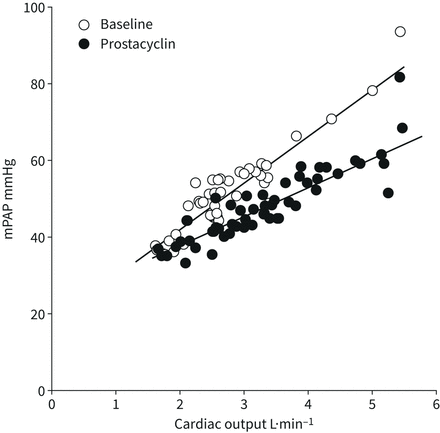
7例患者在接受前列环素治疗6周前后,平均肺动脉压(mPAP)作为心排血量的函数通过运动增加,这与6分钟步行距离平均提高80米有关。前列环素不影响静息肺血管阻力(PVR),但降低运动PVR和mPAP作为心排血量函数的斜率(数据来自[36])。
如图1和3., PAH map -心排血量关系线性调整的斜率小于PVR方程预测[34,35]。这是由于肺血管闭合压力增加[32,33]。运动性心力衰竭肺动脉高压患者map -心排血量的斜率比PVR方程预测的要陡[36]。如前所述[35],这可以解释为运动引起的肺血管收缩和PAWP增加。
在肺血管完全招募的健康个体中,PVR随心排血量增加而降低是由阻力性血管扩张所解释的在体外实验为直径每mmHg变化2%透壁压力[37]。因此,PVR方程可以通过加入阻性血管膨胀系数α(%·mmHg)来改善−1)用R0按mPAP与心排血量之比计算(问”)(38] 该方程允许从一组肺血管压力和流量测量中计算α [37]。导出的膨胀因子α通常为1.5-2%·mmHg−1;男性比绝经前女性低;随着年龄的增长和慢性(但不急性)缺氧暴露而减少[37,39,40]。在早期或潜伏性肺血管疾病中降低[41,42]和保留射血分数(EF)的心力衰竭[42,43]。在PAH中,map -心排血量关系符合线性或准线性模型,因此流量引起的PVR下降主要是由封闭的阻力血管重新招募引起的[35]。如图4,在正常全招募肺中,肺血管扩张仅为1-2%·mmHg−1显著限制运动时心排血量增加时PAP的增加[40,41]。
该方程允许从一组肺血管压力和流量测量中计算α [37]。导出的膨胀因子α通常为1.5-2%·mmHg−1;男性比绝经前女性低;随着年龄的增长和慢性(但不急性)缺氧暴露而减少[37,39,40]。在早期或潜伏性肺血管疾病中降低[41,42]和保留射血分数(EF)的心力衰竭[42,43]。在PAH中,map -心排血量关系符合线性或准线性模型,因此流量引起的PVR下降主要是由封闭的阻力血管重新招募引起的[35]。如图4,在正常全招募肺中,肺血管扩张仅为1-2%·mmHg−1显著限制运动时心排血量增加时PAP的增加[40,41]。
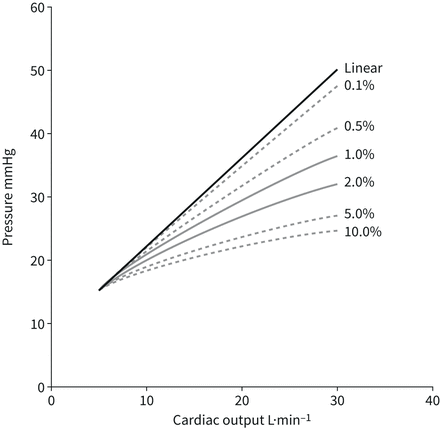
模拟动态运动时的平均肺动脉压(mPAP) -心排血量关系,扩张系数α从0增加到每mmHg透壁压血管直径的10%。α的正常值在1%·mmHg之间−1和2%·毫米汞柱−1.肺血管扩张显著降低高水平心排血量时的mPAP。转载自[40得到许可。
血液不是牛顿流体,因此PVR随血液黏稠度呈线性增加,而血液黏稠度又与红细胞压积呈指数相关。如图5,高血细胞压积、PVR和PAWP时,红细胞压积对PVR的影响增加[44]。贫血或红细胞增多症患者的PVR测量可能明显低估或高估肺血管阻塞。
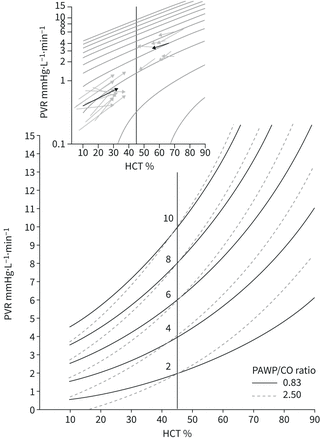
肺血管阻力(PVR)与红细胞压积(HCT)的关系。Iso-PVR曲线可以预测HCT在贫血或红细胞增多患者的PVR“正常化”为45%,在正常或增加肺动脉楔形压(PAWP)时。与心输出量(CO)关系。上方窗口的箭头显示了因输血或血液稀释导致HCT升高或降低的贫血或红细胞增多患者的PVR测量值,平均反应(粗箭头)显示与预测变化相符。转载自[44得到许可。
肺循环的扩张性模型允许根据给定的mPAP和心排血量测量值重新计算肺血管梗阻量[45]。这在图6.心输出量为5l·min−1, mPAP为25 mmHg(以前的肺动脉高压定义)对应50%的梗阻(或切除一个肺),而mPAP为20 mmHg(肺动脉高压的新定义)对应20 - 25%的梗阻。
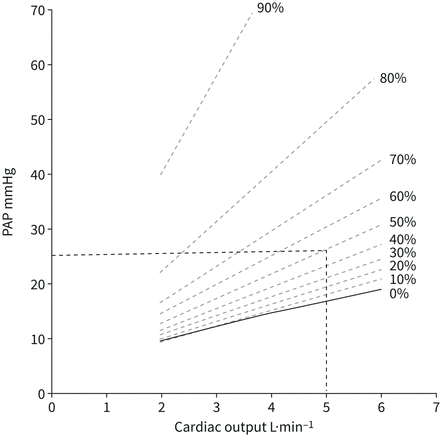
通过线性化调整平均肺动脉压(mPAP)作为心排血量的函数对肺血管阻塞百分比的模型预测。mPAP 25 mmHg或20 mmHg,心排血量为5 L·min−1分别对应50%和25%的梗阻。转载及修改自[45得到许可。
临床意义
通过多点肺血管压力-流量关系对PVR进行细化定义,可能有助于诊断早期肺血管疾病,并有助于了解临床建立的PAH干预措施的影响。贫血或红细胞增多症患者需要纠正红细胞压积。目前定义的正常心输出量PH值对应于肺血管重构和25-50%的血管梗阻。
PVR分区
肺动脉球囊阻塞后测量PAWP的压力衰减曲线呈现出一个较快的部分,对应于上游的动脉阻力,以及一个较慢的部分,对应于包含小动脉、毛细血管和小静脉的下游阻力[46]。这在图1.上游PVR仅在近端CTEPH中增加。PAH下游PVR升高与肺静脉闭塞性疾病(PVOD) [46]。
通过动脉闭塞划分PVR可以确定近端CTEPH患者,他们最有可能受益于手术或介入性去梗阻[47- - - - - -49]。由于肺血管重构的重叠,分析对PAH和PVOD的鉴别没有帮助[4,50]。
临床意义
右心导管插管时单球囊阻塞压力衰减曲线可揭示近端肺动脉阻塞(近端CTEPH)与远端小动脉、毛细血管和或静脉阻塞(PAH, PVOD)之间PVR分配的差异。
肺循环:搏动血流动力学
肺血管阻抗
RV的液压功或“动态后载荷”由阻力、顺应性和反射波之间的相互作用决定[51]。这些肺动脉血流阻力的决定因素可以通过PAP和血流波的频谱分析来量化[7,52,53]。结果图形表示为肺血管阻抗(PVZ)谱或PAP/流量(问')和相位角随频率(图1和7).0 Hz阻抗(mPAP/问’)主要由小血管阻力和左房压决定。随着频率的增加,阻抗受到更多近端动脉阻力的影响。低频相位角为负表明流动导致压力。在高频时,PAP/问的比值减小,并在一个常数值附近振荡,称为特征阻抗(Zc).这是可以计算的Zc在时域上为PAP/的早期收缩斜率问”(54]。Zc是惯性与顺应性的比率,因此可以测量近端肺动脉僵硬程度。
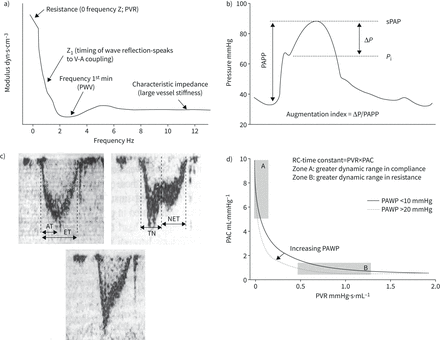
a)肺血管阻抗谱表示为肺动脉压流比(“模量”)作为频率的函数。有关问题的讨论,见图上的说明和文本中的评论。b)严重肺动脉高压的肺动脉压(PAP)曲线显示增加指数增加,或收缩期PAP (sPAP)与上冲程压力(P我)除以PA脉冲压力(PAPP)。c)正常受试者(左图)和严重PH患者(右图和下图)的多普勒肺动脉血流波。肺动脉高压与射血时间(ET)校正后的加速时间(AT)和收缩中期血流减速(缺口)或收缩晚期血流减速相关。TN:开槽时间,NET:开槽到弹射结束的时间。d)高或正常肺动脉楔压(PAWP)患者肺动脉顺应性(PAC)与肺血管阻力(PVR)的双曲线关系。高PAWP会使关系向左移动,同时减少rc时间。转载自[53]、[64]和[77]具有权限。
肺血管阻抗测定已在少数儿科和成人IPAH患者中报道过[55- - - - - -58]。总的模式是PVZ谱向上移位,PAP-flow模量的第一个最小值和最大值移到更高的频率,增加Zc且低频相位角较多为负。增加Zc与rv -动脉解耦有关[57]以及在儿童肺动脉高压患者中比单纯PVR预测的临床结果更差[58]。
临床意义
PAH中PVZ的测量可以评估RV后载荷,但受限于频域分析的复杂性。PVZ决定因素的临床相关性Z0和Zc仍在调查中。
时域脉动血流动力学
严重肺动脉高压的特征是肺动脉脉压升高(PP,或收缩期PAP (sPAP)减去舒张期PAP (dPAP))、收缩期后期压力达到峰值和压力波上冲程的短暂平台期[53]。这些方面由肺动脉顺应性降低(或stroke volume (SV)除以PP)和第一反射压力波延迟增加决定[59]。波反射可以用所谓的增强指数来量化,即M最初提出的sPAP减去平台压力除以PPurgoet al。[60]用于体循环。据报道,与PAH中远端小血管梗阻相比,cteh中近端肺动脉PP增加[61]。然而,基于PP或Murgo指数的这两种情况之间的区别,在个别基础上是不可能的[62]。
肺动脉高压的另一个特点是收缩期晚期或中期血流减慢,也称为“缺口”[63,64]由于第一个反射流波较早返回[59]。通过测量从流波开始到最大收缩中期减速时刻的“到缺口的时间”来量化缺口已被提出作为波反射增加的指标,因此主要是近端CTEPH [65]。但该入路尚未被接受,可能与CTEPH中高压多环芳烃的重叠及远端小血管疾病的共同并存[66]。肺动脉流量缺口的存在与PVR增加的可能性很大相关[67]。
临床意义
人们对PAP和流波形态分析用于肺动脉高压的诊断工作很感兴趣。高脉压、收缩期后期压力峰值和收缩期中期流量减慢是晚期肺血管疾病和/或近端肺动脉梗阻的指标。近端肺动脉硬化增加Zc,可以在时域和频域进行评估,可能与RV后载荷有关。
肺循环的时间常数
肺动脉顺应性主要(>80%)分布于动脉树的外周部分[68],而全身动脉顺应性主要(>80%)在胸主动脉近端[69]。在体循环中,阻力与顺应性无关,但肺动脉顺应性与PVR的乘积(也称为RC-time)保持不变,并在肺动脉高压的严重程度、病因和治疗范围内保持双曲线关系[68- - - - - -73]。这个性质有两个结果。首先,当PAP和PVR仅小幅升高时,肺动脉顺应性成为RV后负荷比PVR更重要的决定因素[73]。二是总水力负荷(W合计)是一个常数分数,约为23% [74因此,W合计可由定流液压载荷(W圣)或按SV计算的mPAP的乘积: 与肺动脉近端梗阻相关的rc时间减少,如实验性肺动脉带或CTEPH [75,76]或增加心衰患者的PAWP [77]。在这些情况下,肺动脉顺应性随PVR的增加而不成比例地降低(图7),因此对RV后载荷的贡献比例更大。
与肺动脉近端梗阻相关的rc时间减少,如实验性肺动脉带或CTEPH [75,76]或增加心衰患者的PAWP [77]。在这些情况下,肺动脉顺应性随PVR的增加而不成比例地降低(图7),因此对RV后载荷的贡献比例更大。
在肺动脉顺应性和PVR的测量中,rc时间是一个很小的数字,会受到误差放大的影响,因此它可能会有很大的变化[78]。然而,报道的rc时间的极端变化对总功的振荡分量影响不大[79]。
有人提出,肺循环的rctime的稳定性解释了为什么sPAP、dPAP和mPAP在正常人和所有可能病因和严重程度的肺动脉高压患者中密切相关[80]。已多次证明sPAP/mPAP和mPAP/dPAP比值非常稳定,实际上符合黄金比例[81]。mPAP可由sPAP通过公式[82]: sPAP和mPAP之间的恒定比例在临床实践中是有用的,因为sPAP很容易通过多普勒超声心动图测量的三尖瓣反流的最大速度来无创地确定[83]。
sPAP和mPAP之间的恒定比例在临床实践中是有用的,因为sPAP很容易通过多普勒超声心动图测量的三尖瓣反流的最大速度来无创地确定[83]。
临床意义
肺动脉顺应性与PVR之间的紧密双曲线关系解释了在轻中度PAH中肺动脉顺应性对后负荷的相对更重要的贡献,从无创估计sPAP预测mPAP的稳定性,以及波反射对RV后负荷的无影响或有限影响。
右心室功能
Ventriculo-arterial耦合
尽管左右心室在胚胎学和结构上有明显的差异,但它们的力学特性是相似的[84]。所谓的“心灵法则”同样适用于两者[85]。心室对后负荷增加的直接适应是根据斯特林心脏定律增加的维度。然而,这种“异质”反应在几分钟内就会被“同量”的收缩性增加所取代,这是遵循Anresp心脏定律的,允许在不扩张和充盈压力增加的情况下保持血流输出。心室肥厚可加强长期的同量适应。当同计量适应耗尽时,异计量适应会再次启动以保持心排血量,但代价是心室扩张和上游充血[6,7,85,86]。
这些概念与通过压力-体积(PV)关系分析左心室(LV)功能是由S佐治亚大学等.[87)。随着人们对右心室功能作为PAH症状和结局的主要决定因素的兴趣的增加,这些研究已经重新兴起[6,7,53,86]。
测量完整心室收缩力的金标准是最大或收缩末期弹性(E西文),或收缩末期压(ESP)与收缩末期容积(ESV)之比。集总后载荷参数可以用ESP和SV的比值来定义。
 因此,RV收缩力与后载荷的耦合关系可以用E西文/E一个比(6,7,53,85,86]。数学模型和实验研究已经证实E西文/E一个在能量消耗最小的情况下,允许SV弹射的最佳比率为1.5至2.0 [85]。
因此,RV收缩力与后载荷的耦合关系可以用E西文/E一个比(6,7,53,85,86]。数学模型和实验研究已经证实E西文/E一个在能量消耗最小的情况下,允许SV弹射的最佳比率为1.5至2.0 [85]。
左心室压力-容积(PV)环呈方形,易于辨认E西文在它的左上角[87]。正常右心室PV环路呈圆形三角形,早期收缩压达到峰值E西文更难辨别的是(图5) [88]。房车的准确定义E西文/E一个比率要求在多个水平的预负荷下,通过操纵系统静脉回流来降低PV回路[88],或单拍PV回路,并分析RV压力曲线[89]。单拍方法依赖于对最大压力的估计(P马克斯)从RV压力曲线的早期和晚期等体积部分的非线性外推。右心室最大压力对应于舒张末期容积(EDV)时非喷射搏动产生的压力(图8).
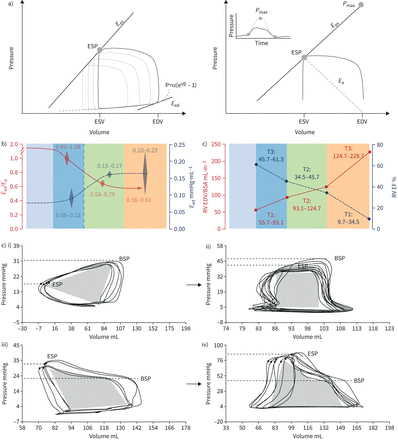
a)计算右心室(RV) -动脉耦合的多拍和单拍方法。在这两种方法中,动脉弹性(E一个)由收缩压(ESP)与冲程容积(SV)之比计算。收缩末期弹性(E西文)作为最大弹性的近似值,由ESP与收缩末期容积(ESV)的比值来估计。在多拍法中,E西文定义为在静脉回流减少时确定的一系列环路的切线。在单拍法中,最大压力(P马克斯)是由右心室压力曲线早期收缩期和舒张期部分的非线性外推估计的E西文由画的直线定义的P马克斯切线到RV的压力相对于体积变化的关系。舒张刚度(β)通过拟合非线性指数计算,p=α (eVβ−1),到舒张期开始和结束时测量的压力和容积。b) RV从正常到逐渐加重的肺动脉高压(PAH) (T1到T3)的演变E西文/E一个,射血分数(EF)舒张末期弹性(E艾德)和右心室舒张末期容积(EDV)校正体表面积(BSA)。右心室扩张时EDV高于正常限度E西文/E一个EF分别近似于0.8和0.35。增加E艾德镜子下降E西文/E一个.c)从无肺动脉高压患者i)到重度肺动脉高压患者iv)的RV压力-容积环路,呈进行性变化,向收缩期后期压力峰值和“缺口”转移,而ESP高于射血起始压力(BSP)。转载自[86]、[96]和[99得到许可。
右心室功能与肺循环的耦合E西文和E一个2004年首次报道在6名PAH患者中进行测量[90]。研究表明E西文大约是对照组的三倍,但降低了E西文/E一个指示RV故障的比率。这些结果已在小组PAH或cteh患者中得到证实[91- - - - - -94]。在这些研究中,E西文/E一个采用单拍法[90,92,93]或降低静脉回流的PV循环家族[91,94]。收缩末期弹性不断增加[90- - - - - -94]。的E西文/E一个比率在静止时维持或降低[90- - - - - -94],但在运动时持续减少[93,94]。减少E西文/E一个同时伴有EDV的增加,这与运动引起的右心室衰竭相对应[94]。
测量E西文/E一个单拍或多拍比率法平均产生相同的结果,但有个别差异[95]。右心室功能与高血压肺循环的耦合有保留:右心室尺寸增大时E西文/E一个Ratio降至0.7-0.8的临界值,因此约为半正常[96) (图8).在PH值相似的情况下E西文/E一个与男性多环芳烃患者相比,女性患者的多环芳烃保存更好[97]。的E西文/E一个比值可独立预测PAH预后[95]。
如图8时,右心室PV环的形状随肺动脉高压的进展而变化,由无PH值受试者的收缩早期压力峰值的三角形形状,变为收缩后期压力峰值和收缩中期压力下降的梯形和三角形形状(“切口”)[98,99]。右心室PV环的这些形态学改变已被证明与预后相关[99]。
临床意义
右心室功能对PAH后负荷增加的适应依赖于收缩力的增加以匹配动脉弹性的增加。PAH中的rv -动脉偶联在运动时减少,在休息时保持或减少。右心室动脉解耦最终导致右心室扩张,右心室充盈压力增加,临床表现为右心衰。动脉-动脉耦合与预后相关。
床边rv -动脉耦合的简化测量
自E西文/E一个比率有压力作为一个常用术语,它可以简化为体积之比[One hundred.,101]:
 SV/EDV比值的临界值理论上为0.54[,低于该临界值,RV维数增加到正常上限之上,同时保留SV]。102]。由于SV等于EDV−ESV,因此
SV/EDV比值的临界值理论上为0.54[,低于该临界值,RV维数增加到正常上限之上,同时保留SV]。102]。由于SV等于EDV−ESV,因此 这相当于35%的RVEF [102]
这相当于35%的RVEF [102]
因为E西文/E一个比率有内置的通用体积项,它也可以简化为压力的比率[101,103]:
 根据这些方程,RVEF可由RV压力曲线导出[104]:
根据这些方程,RVEF可由RV压力曲线导出[104]: rv -肺动脉耦合的简化测量存在局限性[105]。体积法假设E西文作为一条穿过原点的直线,这是不现实的,因为心室收缩末期弹性曲线略呈曲线,与容积轴的正外推确定无应力容积(V0) [106]。压力法要求的计算P马克斯,已报道了几种方法,但这些方法可能并不等效[89,93,101,104]。有人建议在右心导管插入术中用更容易测量的mPAP取代ESP [103]。然而,mPAP低估了ESP与PAP增加的比例[107]根据方程:
rv -肺动脉耦合的简化测量存在局限性[105]。体积法假设E西文作为一条穿过原点的直线,这是不现实的,因为心室收缩末期弹性曲线略呈曲线,与容积轴的正外推确定无应力容积(V0) [106]。压力法要求的计算P马克斯,已报道了几种方法,但这些方法可能并不等效[89,93,101,104]。有人建议在右心导管插入术中用更容易测量的mPAP取代ESP [103]。然而,mPAP低估了ESP与PAP增加的比例[107]根据方程: mPAP和ESP之间的压力依赖差异与PV环形状的变化有关[98,99如:图8.
mPAP和ESP之间的压力依赖差异与PV环形状的变化有关[98,99如:图8.
RVEF是PAH中最有效的rv -肺动脉耦合的简单影像学评估方法。多项研究表明,RVEF是PAH预后的独立预测因子,其临界值为35-40%,比正常值低约60-70% [108- - - - - -110]。一项大规模研究表明,RVEF <37%、37-54%和>54%与PAH高、中、低生存率降低风险显著相关[111]。一项荟萃分析估计,RVEF每降低1%,2年临床恶化的风险就会增加5%,5年死亡的风险就会增加2%(数字四舍五入)[112]。由二维超声心动图评估的右心室舒张末期和收缩末期面积之间的差异与舒张末期面积的比值(FAC)作为RVEF的替代,也被证明是预后的预测指标,同样严格确定的临界值为~ 35% [113]。
通过靶向治疗逆转RV尺寸并增加EF或FAC可使PAH预期寿命恢复正常[114]。改善EF或FAC的反向重塑最好通过PVR降低> 40-50% [114- - - - - -117]。PVR的这种降低需要至少两种针对不同途径的治疗方法的联合,而肠外前列环素可能更一致地实现[114- - - - - -117]。
临床意义
PAH严重的右心室-动脉解耦与右心室扩张、全身充血和预期寿命缩短有关。当EF或SV/ESV分别下降至35%和54%以下时,右心室尺寸增加即为右心衰,其变异性与卒中量保持或减少有关。右心室的反向重塑可以改善PAH的预后,并通过联合治疗将PVR降低至少40-50%。
舒张功能
右心室功能与后负荷的耦合具有不可避免的舒张成分[6,85,86,96,101,118,119]。如图8,舒张功能由舒张弹性曲线描述,该曲线可由可变负荷下的一系列压力-容积循环或通过在单个PV循环上拟合收缩末期和舒张末期压力-容积关系来确定[86]。拟合由非线性指数曲线组成,通过原点和收缩末期和舒张末期的压力-体积坐标与公式Pe =α(Vβ−1),其中α为曲线拟合常数,β为舒张刚度常数[86]。
有数据,但来自有限数量的患者,表明舒张刚度常数β独立预测PAH的结局[118,119]。这需要确认。多环芳烃舒张适应后负荷的生物学正在探索中[118]。通过简单的舒张末期压力-容积坐标简单量化的舒张刚度保留了更复杂的β计算的预测能力[119]。PAH的收缩和舒张硬度都增加,当PVR通过靶向治疗的作用充分降低时,PAH的收缩和舒张硬度随着右心室体积的减小而降低[120]。
临床意义
PAH时右心室舒张和收缩弹性均增加。目前正在探索舒张功能障碍的生物学及其与多环芳烃预后的相关性。
室的相互作用
右心室功能必须在其与左心室功能直接和间接相互作用的背景下加以理解[84,121]。舒张期心室相互作用阴性(即.由于心室争夺空间,导致一个心室舒张功能改变而改变另一个心室舒张功能)发生在PAH中。这改变了左室充盈,最终可能与左室收缩功能下降、左室萎缩重塑和心排血量受损有关[121]。由于左室收缩力降低,收缩期心室负相互作用可能增加舒张期相互作用的影响[121]。实验表明,主动脉收缩通过同程适应增强左室收缩,显著改善肺动脉带带动物的右室功能[122]。在完整的实验动物心脏中,左室收缩压的20-40%来自左室收缩,左室收缩压的4-10%来自右室收缩[123]。
收缩期心室相互作用本质上是由直接的机械夹带效应解释的,因为两个心室都有共同的室间隔,并且被共同的纤维包围[84,121]。另一个因素是全身性低血压时冠状动脉灌注压降低,这可能导致右心室相对缺血,随后与肺循环脱钩,但对左心室收缩的敏感性增加[124]。右心室充盈压力升高和血压过度降低共同导致右心室缺血和收缩力下降[125]。
心室相互作用在急性右心衰(如肺栓塞)或终末期PAH右心衰中起重要作用。纠正舒张和收缩期心室负相互作用是这些患者治疗的重要组成部分,因为他们在重症监护室住院[126]。
影像学研究揭示的心室负相互作用的另一个原因是收缩后收缩或“缩短”的区域和室间不同步,已被证明与肺动脉压力增加并行发展,并导致右心室收缩功能改变和左室充盈不足[127]。右心室局部不同步(心室间收缩同步性丧失)和非同步性(心室局部收缩同步性丧失)现在可以通过散斑跟踪超声心动图来识别和量化[128,129]。右心室收缩的非同步性可能已经出现在早期或PH值边缘[129]。晚期PAH患者右心室收缩的非同步化与功能状态变差和生存率降低有关[128]。
临床意义
负舒张和收缩相互作用是右心衰rv -动脉耦合的主要决定因素。心室内和心室间收缩同步性的丧失导致心室-动脉耦合改变,并与缩短生存率有关。
右心室受压
压迫右心室测量其“收缩储备”可能揭示与肺循环的交界或潜在功能解耦[95]。
多普勒超声心动图通过三尖瓣反流的最大速度估计运动诱导的右心室收缩压升高,已被证明是PAH或CTEPH患者生存的一个强有力的预测因素[130]。然而,单一的压力估计并不能充分描述rv -肺动脉耦合[94]。另一种方法是评估运动引起的心排血量增加[131]。自E西文/E一个比值和EF是相关的,在运动中保持或增加[93,94,132],运动压力EF应该是一个有效的挑战,以发现早期或未决的RV失败。这种方法在历史上曾使用放射性核素血管造影来识别COPD的右心室功能障碍[133]。然而,磁共振成像测量运动中的右心室体积在技术上具有挑战性,而且通常不可用。
正在考虑用低剂量多巴酚丁胺代替运动应激,并通过三尖瓣环面收缩偏移(TAPSE)或三尖瓣环空S波测量右心室收缩反应的可能性[134]。有实验工作表明多巴酚丁胺诱导的右心室收缩功能这些指标的升高反映了右心室-动脉耦合的静息状态[135]。
最大摄氧量的量度(V”O2马克斯)可被视为间接右心室压力测试,因为PAH有氧能力的主要决定因素是由右心室-左心室耦合决定的最大心排血量[134]。然而,V”O2马克斯与心力衰竭一样,多环芳烃的发病率也受到心外因素的影响,包括神经体液变化以及肌肉对流和扩散氧运输机制的匹配[15]。
临床意义
对右心室进行动态测试以评估其收缩储备,可能揭示右心室与肺循环的潜在或边缘性解耦。对右心室施加应力的最佳方法尚未确定。
的角度来看
由于右心室功能是多环芳烃症状和结局的主要决定因素,因此正在开发简单的非侵入性评估方法。其中之一是TAPSE(与估计的比值)E西文)到sPAP(以估计后负荷),这些指标在标准多普勒超声心动图中很容易测量[136]。TAPSE/sPAP比值已在侵入性测量中得到验证E西文/E一个比(137,138]并被证明与心力衰竭的预后相关[136,137]和多环芳烃[139]。
然而,多环芳烃的生理基础不仅仅是右心室功能障碍。PAH患者的管理需要全面了解肺力学、肺气体交换、肺血管功能以及右心与肺循环耦合的基本生理学。这一知识不仅对于正确评估治疗干预措施至关重要,而且对于评估快速增长的对这种疾病的细胞和分子生物学的理解的临床相关性至关重要。
可共享的PDF
脚注
利益冲突:R. Naeije报告了与AOP Orphan Pharmaceuticals、Johnson & Johnson、Lung Biotechnology Corporation和United Therapeutics的关系,包括咨询、演讲费和顾问委员会成员。
利益冲突:M.J. Richter获得了来自德国研究基金会(DFG, 413584448)和合作研究中心(SFB) 1213 -肺动脉高压和肺心病(资助号SFB1213/1,项目B08;德国研究基金会,波恩,德国)。
利益冲突:鲁宾在Actelion、SoniVie、Gossamer Bio和Bellerophon担任顾问。
支持声明:M.J. Richter获得了JLU-CAREER计划(德国研究基金会,DFG, 413584448)和合作研究中心(SFB) 1213 -肺动脉高压和肺心病的资助,资助号为SFB1213/1,项目B08(德国研究基金会,波恩,德国)。
- 收到了2021年8月25日。
- 接受2021年10月18日。
- 版权所有©作者2022。
本版本根据知识共享署名非商业许可4.0的条款发布。为商业复制权利和权限联系权限在}{ersnet.org











![Progressive remodelling of the pulmonary circulation (upper row) and of the right heart (lower row) with, in-between, typical measurements of pulmonary ventilation and gas exchange, steady-flow and pulsatile-flow haemodynamics and right ventricular (RV) function in pulmonary arterial hypertension (PAH). Vascular remodelling was defined by microscopic views from normal (left) to medial hypertrophy, thickening of the three layers of vascular wall and plexiform lesions (upper right). Cardiac remodelling was defined by two-dimensional four-chamber echocardiographic views of right atrial and ventricular dilatation and septal shift ventricular dilatation, respectively. Arrows indicate hypothetical disease progression with increasingly dilating RV and right atrial dimensions. In between are measurements in typical patients with severe idiopathic PAH (IPAH): a) quasi-normal distribution of alveolar ventilation (V′A) and perfusion (Q′) as a function of V′/Q′ relationships, arterial partial pressure of oxygen (PaO2) and physiological dead space (dead space volume (VD)/tidal volume (VT)), but with increased V'A; b) increased ventilation (V′E) as a function of carbon dioxide output (V′CO2) during exercise; c) Poon-adjusted mean pulmonary artery pressure (PAP) as a function of cardiac output (Q′) increased by either exercise or dobutamine showing a decreased slope and increased extrapolated pressure intercept (n=7 patients); d) PAP decay curve after single arterial occlusion showing increased inflection point; e) RV pressure curve with increased pulse pressure, late systolic peaking and increased augmentation index; f) Doppler RV outflow tract flow with shortened acceleration time and mid-systolic deceleration; g) pulmonary artery impedance spectrum, or pressure/flow and phase as a function of frequency, showing an upward shift with increased 0 Hz and high-frequency impedances and low-frequency negative phase angle, with increased characteristic impedance (Zc) in either frequency- or time-domain; h) RV pressure–volume loops with increased end-systolic elastance (Ees) in the presence of increased arterial elastance (Ea), but decreased Ees/Ea and increased dimensions as compared to control measurements. PAPP: pulmonary arterial pulse pressure; sPAP: systolic pulmonary arterial pressure; Pi: inflection of the upstroke pressure. Histological views and echocardiographic images are courtesies from Andrew Peacock (Regional Heart and Lung Centre, Glasgow, UK) and Michele D'Alto (Monaldi Hospital, Naples, Italy). Panel c) is reproduced from [34], with permission.](http://www.qdcxjkg.com/content/erj/59/6/2102334/F1.large.jpg?width=800&height=600&carousel=1){kind=link}
{kind=link}
![a) Distribution of arterial blood gases in 243 patients with idiopathic pulmonary arterial hypertension (IPAH). Most values are decreased, with 51% of PaO2 <70 mmHg, and 45% of PaCO2 <33 mmHg taken as lower limits of normal (shaded columns). b) Alveolar ventilation (V′A)/perfusion (Q′) distributions in a patient with IPAH before and after a normalisation procedure correcting for increased V′A and decreased Q′, and in a middle-aged control (from left to right). In the IPAH patient, V′A/Q′ distributions are shifted to higher V′A/Q′, but V′A and Q′ modes remain matched. Both the IPAH patient and the healthy control have a small amount of perfusion to low V′A/Q′. The IPAH patient has also a small amount of shunt (V′A/Q′=0) which was increased with correction for high V′A and low Q′. c) Ventilation (V′E) as a function of carbon dioxide (CO2) output adjusted for interindividual variability in 12 PAH patients and in 10 controls. V′E was increased at rest and more so at exercise. d) Plotting V′E/V′CO2 as a function of arterial carbon dioxide tension (PaCO2) at the anaerobic threshold during exercise (AT) uncovers increased dead space ventilation (dead space volume (VD)/tidal volume (VT)) and chemosensitivity as causes of increased ventilation in PAH and COPD, or only increased chemosensitivity in heart failure patients. The arterial to end-tidal oxygen tension gradient increased >5 mmHg (the upper limit of normal) in PAH and in COPD. Reproduced from [16] and [25] with permissions.](http://www.qdcxjkg.com/content/erj/59/6/2102334/F2.large.jpg?width=800&height=600&carousel=1){kind=link}
{kind=link}
![Mean pulmonary artery pressure (mPAP) as a function of cardiac output increased by exercise in seven patients before and after 6 weeks of prostacyclin therapy, which was associated with an improvement in the 6-min walk distance by an average of 80 m. Prostacyclin did not affect resting pulmonary vascular resistance (PVR), but decreased exercise PVR and the slope of mPAP as a function of cardiac output (data from [36]).](http://www.qdcxjkg.com/content/erj/59/6/2102334/F3.large.jpg?width=800&height=600&carousel=1){kind=link}
{kind=link}
![Modelled mean pulmonary arterial pressure (mPAP)–cardiac output relationships during dynamic exercise with progressively increased distensibility coefficients α from 0 to 10% of vessel diameter per mmHg transmural pressure. Normal values for α are between 1%·mmHg−1 and 2%·mmHg−1. Pulmonary vascular distension markedly decreases mPAP at high levels of cardiac output. Reproduced from [40] with permission.](http://www.qdcxjkg.com/content/erj/59/6/2102334/F4.large.jpg?width=800&height=600&carousel=1){kind=link}
{kind=link}
![Pulmonary vascular resistance (PVR) as a function of haematocrit (HCT). Iso-PVR curves allow for a prediction of PVR “normalised” for HCT at 45% in anaemic or polycythaemic patients, at normal or increased pulmonary artery wedge pressures (PAWP) versus cardiac output (CO) relationships. Measured PVR in anaemic or polycythaemic patients at increased or decreased HCT by blood transfusion or haemodilution are shown by arrows in the upper window, with mean responses (thick arrows) showing conformance to predicted changes. Reproduced from [44] with permission.](http://www.qdcxjkg.com/content/erj/59/6/2102334/F5.large.jpg?width=800&height=600&carousel=1){kind=link}
{kind=link}
![Model predictions of percentage of pulmonary vascular obstruction by linearised adjustments of mean pulmonary artery pressure (mPAP) as a function of cardiac output. mPAP 25 mmHg or 20 mmHg at a cardiac output of 5 L·min−1 correspond to 50% and 25% obstruction, respectively. Reproduced and modified from [45] with permission.](http://www.qdcxjkg.com/content/erj/59/6/2102334/F6.large.jpg?width=800&height=600&carousel=1){kind=link}
{kind=link}
![a) Pulmonary vascular impedance spectrum expressed as pulmonary artery pressure–flow ratio (“modulus”) as a function of frequency. For discussion of the relevant points, see indications on figure and comments in the text. b) Pulmonary artery pressure (PAP) curve in severe pulmonary hypertension showing an increased augmentation index, or gradient between systolic PAP (sPAP) and inflection of the upstroke pressure (Pi) divided by PA pulse pressure (PAPP). c) Doppler pulmonary artery flow waves in a normal subject (left panel) and in patients with severe PH (right and lower panels). Pulmonary hypertension was associated with a decreased acceleration time (AT) corrected for ejection time (ET) and mid-systolic deceleration of flow (notching) or late systolic deceleration of flow. TN: time to notching, NET: time from notching to end-ejection. d) Hyperbolic relationship between pulmonary artery compliance (PAC) and pulmonary vascular resistance (PVR) in patients with either a high or a normal pulmonary artery wedge pressure (PAWP). A high PAWP shifts the relationship to the left with decreased RC-time. Reproduced from [53], [64] and [77] with permissions.](http://www.qdcxjkg.com/content/erj/59/6/2102334/F7.large.jpg?width=800&height=600&carousel=1){kind=link}
{kind=link}
![a) Multiple- and single-beat methods for calculating right ventricular (RV)–arterial coupling. In both methods, arterial elastance (Ea) is calculated from the ratio of end-systolic pressure (ESP) to stroke volume (SV). End-systolic elastance (Ees) as an approximation of maximum elastance is estimated by the ratio of ESP to end-systolic volume (ESV). In the multiple-beat method, Ees is defined by a tangent to a family of loops determined at decreasing venous return. In the single-beat method, a maximum pressure (Pmax) is estimated from the nonlinear extrapolation of the early systolic and diastolic portions of the RV pressure curve, and Ees defined by a straight line drawn from Pmax tangent to RV pressure to relative change in volume relationship. Diastolic stiffness (β) is calculated by fitting the nonlinear exponential, p=α (eVβ−1), to pressure and volume measured at the beginning and end of diastole. b) Evolution from normal to progressively more severe pulmonary arterial hypertension (PAH) (T1 to T3) of RV Ees/Ea, ejection fraction (EF) end-diastolic elastance (Eed) and RV end-diastolic volume (EDV) corrected for body surface area (BSA). The RV dilates with EDV above limits of normal when Ees/Ea and EF approximate 0.8 and 0.35, respectively. Increased Eed mirrors decreased Ees/Ea. c) RV pressure–volume loops from patients i) without pulmonary hypertension to iv) severe pulmonary hypertension, showing progressive change in shape with shift to late systolic peaking of pressure and “notching”, while ESP becomes higher than pressure at the onset of ejection (BSP). Reproduced from [86], [96] and [99] with permission.](http://www.qdcxjkg.com/content/erj/59/6/2102334/F8.large.jpg?width=800&height=600&carousel=1){kind=link}
{kind=link}