





文摘
背景Benralizumab人性化,anti-interleukin-5α受体单克隆抗体与anti-eosinophilic活动。缺乏岩藻糖(afucosylation)增加其亲和力CD16a和显著增强锁定细胞自然杀伤(NK)细胞的细胞毒性。尽管benralizumab临床证明有效的临床试验严重哮喘患者和hypereosinophilic综合症,深入描述anti-eosinophilic机制的行动仍然是难以捉摸的。
方法这里,我们进一步调查的机制参与benralizumab anti-eosinophilic活动通过使用相关的主要人类自体的细胞培养和real-time-lapse成像结合流式细胞术。
结果在NK细胞的存在,benralizumab强嗜酸性粒细胞凋亡诱导了细胞色素的上游Caspase-3/7诱导和upregulationc。此外,我们发现了一个以前未被发现的机制,它能让benralizumab引起嗜酸性粒细胞吞噬/ efferocytosis巨噬细胞,这一过程被称为依赖抗体细胞的吞噬作用。使用活细胞成像,我们埃及的逐步过程主要通过激活巨噬细胞嗜酸性粒细胞凋亡和吸收。通过仔细观察细胞共培养的分析,我们发现了一个新角色macrophage-derived肿瘤坏死因子(TNF)进一步加强benralizumab-mediated嗜酸性粒细胞通过激活肿瘤坏死因子受体1的嗜酸性粒细胞凋亡。TNF-induced嗜酸性粒细胞凋亡与细胞色素有关cupregulation,线粒体膜两极化和Caspase-3/7活动增加。此外,激活NK细胞被发现放大这个轴通过分泌interferon-γ,随后驾车由巨噬细胞肿瘤坏死因子表达。
结论我们的数据提供更深入的洞察事件的及时出现导致benralizumab-induced嗜酸性粒细胞凋亡,表明额外的机制可能导致benralizumab的有力anti-eosinophilic活动在活的有机体内。重要的是,afucosylation benralizumab强烈增强其力量的机制研究。
文摘
新见解解释benralizumab的有力anti-eosinophilic活动,一个anti-IL-5Rαafucosylated单克隆抗体与增强耗尽力量严重患者嗜酸性炎症https://bit.ly/3yUnsnn
介绍
积累的组织嗜酸性粒细胞在慢性炎性疾病会导致组织损伤通过释放细胞毒性产品包含在他们的颗粒1,2]。interleukin-5的不可或缺的作用(IL-5)嗜酸性粒细胞发展、分化和激活(3,4)促使免疫抗体疗法的发展破坏IL-5 / IL-5受体(IL-5R)轴减少嗜酸性粒细胞数量和他们在折磨组织激活(5]。事实上,两种单克隆抗体针对IL-5 mepolizumab (Nucala)和reslizumab (Cinqair),和一个针对IL-5Rα,benralizumab (Fasenra),现在批准治疗与嗜酸性粒细胞的表型严重哮喘的治疗,也嗜酸性肉芽肿病与polyangiitis mepolizumab (Churg-Strauss综合征)。在所有三个抗体已被证明减少组织中嗜酸性粒细胞计数的患者不同程度(6,7),他们有不同的作用机制。Mepolizumab reslizumab绑定和抑制IL-5,而benralizumab触发锁定细胞介导细胞毒性(ADCC)通过绑定IL-5Rα嗜酸性粒细胞。我们以前报道,afucosylation寡糖链的CH2地区benralizumab Fc的域导致增强Fcγiii a受体(CD16a)绑定和后续ADCC-mediated嗜酸性粒细胞凋亡在体外在自然杀伤(NK)细胞的存在8]。由于其高效力[8),快行动开始9和明显的嗜酸性粒细胞损耗的病人10),进一步调查机制,可能导致benralizumab anti-eosinophilic活动是很重要的。
为了这样做,我们开发活细胞成像方法结合流式细胞术和生化分析使用主要人类嗜酸性粒细胞和效应细胞在体外,而不是执行在活的有机体内研究老鼠,由于缺少benralizumab大鼠IL-5Rα和Fc受体生物学老鼠和人类之间的差异。在这里,我们表明,benralizumab诱发的Caspase-3/7活化和细胞色素cupregulation嗜酸性粒细胞在NK细胞的存在,进一步充实其pro-apoptotic活动。此外,我们首次展示benralizumab介导嗜酸性粒细胞吞噬/ efferocytosis巨噬细胞,这一过程称为锁定细胞吞噬作用(ADCP),刺激肿瘤坏死因子(TNF)端依赖巨噬细胞细胞毒性。自从TNF此前报道引起嗜酸性粒细胞凋亡通过肿瘤坏死因子受体1 (TNFR1) (11通过巨噬细胞),我们调查benralizumab-induced TNF分泌。我们能够证明增强TNF的表达通过激活巨噬细胞和嗜酸性粒细胞凋亡TNFR1表达增加。TNF的封锁/ TNFR1导致ADCP的嗜酸性粒细胞减少和TNF / TNFR1-mediated嗜酸性粒细胞巨噬细胞的凋亡。NK细胞的存在进一步提高TNFR1-mediated凋亡的嗜酸性粒细胞释放interferon-γ(IFN-γ)。重要的是,观察anti-eosinophilic benralizumab活动明显增强与父母相比fucosylated benralizumab同种型。综上所述,我们的数据提供额外的解释benralizumab有力anti-eosinophilic活动的病人。
方法
一个完整的描述中提供的方法补充材料。
研究批准
所有健康的捐赠者匿名参加阿斯利康研究样本采集程序。这些志愿者报名前提供书面知情同意。一个独立的伦理委员会批准成立的阿斯利康研究样本采集程序和使用标本为研究目的。本研究进行了符合赫尔辛基宣言,国际协调委员会/良好的临床实践指南,适用的监管要求和阿斯利康生物伦理学政策。
结果
以前,我们证明benralizumab-mediated嗜酸性粒细胞凋亡(half-maximal有效浓度(EC50)0.9 pmol·L−1)通过ADCC [8通过量化膜联蛋白V-expressing嗜酸性粒细胞在NK细胞的存在。进一步探索这个最初的观察,我们结合流式细胞术和活细胞成像研究自体人类嗜酸性粒细胞/ NK细胞共培养实验。
通过NK细胞毒性Benralizumab介导caspase-dependent嗜酸性粒细胞凋亡
人类健康的捐赠者与外周血嗜酸性粒细胞,接受抗体,沾染了远红外的脂质膜染料和混合与自体phycoerythrin-Texas Red-labelled NK细胞比例1:3(嗜酸性粒细胞:NK细胞)(补充图S1a)。在6 h,细胞凋亡在CD66b检测+Siglec-8+嗜酸性粒细胞通过流式细胞术和蓝色阳离子核酸染料PO-PRO-1,渗透到等离子体膜在凋亡级联起始。正如所料,在NK细胞的存在,benralizumab显著增强嗜酸性粒细胞死亡而fucosylated anti-IL-5Rα抗体,增加比例的PO-PRO-1就证明了这一点+CD66b+Siglec-8+细胞(图1一个,补充图印地和补充视频S1和S2)。Benralizumab独自没有影响嗜酸性粒细胞缺乏NK细胞死亡(补充图就是S1c)。Benralizumab-induced PO-PRO-1+嗜酸性粒细胞也积极Caspase-3/7活动和细胞色素c与此同时增加(图1 b和c)。与此一致的是观察,半胱天冬酶活动的实时成像证实的CellEvent Caspase-3/7试剂检测之前PO-PRO-1染料在垂死的嗜酸性粒细胞(图1 d和补充视频S3)。
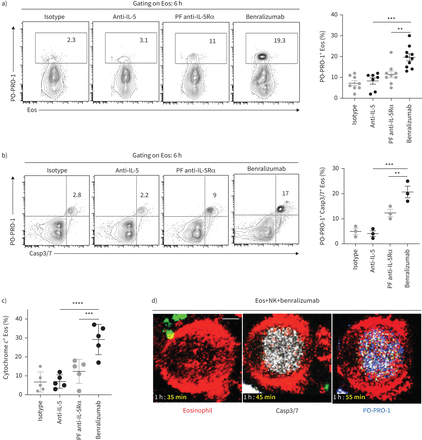
Benralizumab诱发caspase-dependent嗜酸性粒细胞(Eos)由自然杀伤(NK)细胞凋亡。主要人类NK细胞和嗜酸性粒细胞与健康的捐赠者是单独的标签。NK细胞含有LysoTracker绿色有选择地标签NK细胞溶素的颗粒。嗜酸性粒细胞与10 nM的预处理表示抗体,与NK细胞混合后6 h和分析。a - c)嗜伊红血球细胞死亡,评价PO-PRO-1蓝色染料的检测)凋亡和坏死细胞(n = 7), b)半胱天冬酶激活评估CellEvent Caspase-3/7 (Casp3/7)试剂(n = 3),细胞色素c)c(n = 3),表示为CD66b总额的百分比+Siglec-8+嗜酸性粒细胞。数据均值±扫描电镜。d)代表帧选择从实时成像数据,每隔10分钟,显示激活半胱天冬酶的嗜酸性粒细胞CellEvent Caspase-3/7试剂,在白色的。共焦显微镜用于实时成像为90分钟。图片是每5分钟。比例尺:2µm (n = 4)。采用双向方差分析与图基的多重比较。* *:p < 0.01;* * *:p < 0.001;* * * *:p < 0.0001, benralizumab与控件(anti-interleukin (IL) 5或父fucosylated anti-IL-5受体α(PF anti-IL-5Rα))。介绍了统计值补充表S2。
Benralizumab结合NK细胞和嗜酸性粒细胞,把他们在密切接触和促进NK细胞嗜酸性粒细胞凋亡[12]。这些直接NK cell-eosinophil交互可以触发释放专门分泌溶酶体携带溶解性产品,如穿孔素和granzyme B, NK-mediated所需的细胞死亡(13]。一旦激活,NK细胞移植CD137, costimulatory CD16受体(14],lysosome-associated膜蛋白(LAMP-1) / CD107a [15),NK细胞脱粒的一项指标。通过监控这些标记物的表达在ADCC化验,我们观察到显著增加CD137表面表达CD56+CD94+NK细胞相对于父fucosylated anti-IL-5Rα抗体。这个反应是增加LAMP-1密度和生产激活NK细胞的穿孔素和granzyme B brefeldin的存在(图2一个和b)。而在benralizumab更高比例的激活NK细胞显示增加CD137的表达和LAMP-1(33%)、低比例granzyme b和穿孔素呈阳性(14%)(图2一个和b)。一个合理的解释NK颗粒的内部股票6 h后刺激的早期脱粒NK细胞在免疫突触的建立与嗜酸性粒细胞(图2 c和补充视频S3和S4)。
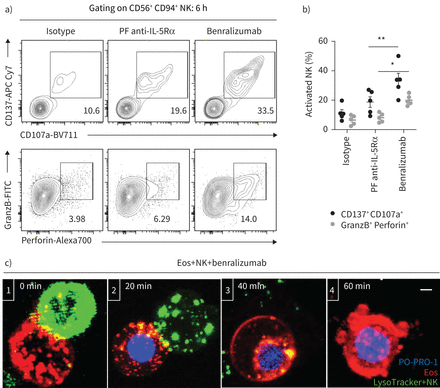
自然杀伤(NK)细胞细胞毒性介导benralizumab-induced嗜酸性粒细胞(Eos)细胞凋亡。主要的嗜酸性粒细胞和NK细胞中描述就做好了准备图1。a、b)改变NK激活(CD137及CD107a)和NK细胞溶解的(granzyme b (GranzB)和穿孔素)标记:a)评估通过流式细胞术和b)表示为CD56总额的百分比+CD94+NK细胞(n = 4)。数据均值±扫描电镜。c)逐步阶段NK-mediated嗜酸性粒细胞benralizumab引起的死亡。细胞活共焦显微镜成像的90分钟。图片是代表帧收购大约每2分钟。1)NK免疫突触之间建立了NK细胞和嗜酸性粒细胞。2)NK细胞溶素的嗜酸性粒细胞的颗粒停靠在细胞接口启动细胞死亡PO-PRO-1进入细胞。3)NK分离的嗜酸性粒细胞分娩后NK细胞溶素的颗粒。4)嗜酸性粒细胞经历凋亡的染色质缩合、膜起泡和核崩溃。µm比例尺:2。采用双向方差分析与图基的多重比较。 *: p<0.05; **: p<0.01, benralizumab与父母fucosylated anti-interleukin-5受体α(PF anti-IL-5Rα)。介绍了统计值补充表S2。
破译细胞相互作用导致嗜酸性粒细胞凋亡诱导benralizumab,我们进行了实时成像的NK cell-eosinophil交互使用共焦显微镜每2分钟2 h。嗜酸性粒细胞和NK细胞co-incubated 1:3的比例在benralizumab PO-PRO-1染料,图像采集前2 h。我们的形象分析证实,细胞刺激benralizumab开始杀戮过程通过NK cell-eosinophil免疫突触的形成(图2 c1),引发颗粒极化(溶解性颗粒的迁移NK细胞表面)(图2 c2)。随后,这些颗粒与嗜酸性粒细胞膜融合,提供细胞毒性介质为嗜酸性粒细胞的胞质(图2 c3),导致细胞膜出泡和核分裂(图2 c4),凋亡的死亡的标志。虽然颗粒迅速分化发生,嗜酸性粒细胞的引发死亡,被PO-PRO-1条目,直到∼20分钟才想起后,随着时间的增加(补充视频S3和S4)。
综上所述,我们的数据支持细胞凋亡的主要机制NK细胞嗜酸性粒细胞死亡benralizumab的存在。
Benralizumab上调嗜酸性粒细胞巨噬细胞所吸收
鉴于巨噬细胞表达CD16a,调解的Fcγ受体介导吞噬作用[16通过分泌细胞毒性机制)和诱导细胞死亡(17,18),我们质疑benralizumab可以通过激活巨噬细胞死亡引起嗜酸性粒细胞。通过结合实时成像和流式细胞术,我们开发了一个新的分析方法来量化嗜酸性粒细胞巨噬细胞所吸收。人类血液CD16+monocyte-derived巨噬细胞被自体培养嗜酸性粒细胞抗体治疗的优化比例1:5 ng在无血清媒体补充50毫升−1巨噬细胞集落刺激因子和成像每5分钟6 h。在这种情况下,我们经常观察个人巨噬细胞能吞噬生活(吞噬作用)(图3一)或凋亡的嗜酸性粒细胞(efferocytosis) (图3 b)。嗜酸性粒细胞死亡的总数被评为吞没了活着的总和(吞噬作用)或PO-PRO-1死了+嗜酸性粒细胞(efferocytosis)后6 h刺激。
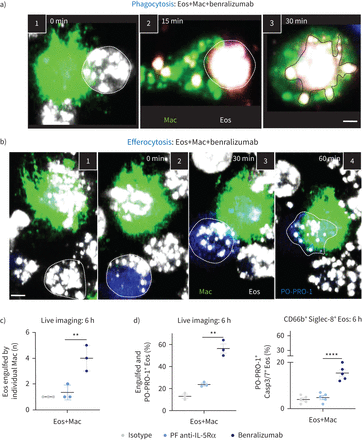
记录的步骤benralizumab-mediated嗜酸性粒细胞(Eos)消耗巨噬细胞(Mac)吞噬作用或efferocytosis。人类主要自然杀伤(NK)细胞和嗜酸性粒细胞与健康的捐赠者是单独的标签。巨噬细胞是沾着绿色的脂质膜染料。嗜酸性粒细胞是预处理的10 nM的benralizumab然后贴上了红色的脂质膜染料。Red-labelled嗜酸性粒细胞与green-labelled co-incubated NK细胞的PO-PRO-1检测细胞死亡。共焦显微镜配合是活细胞成像的90分钟。a)逐步阶段macrophage-mediated嗜酸性粒细胞吞噬benralizumab的目标。1)Macrophage-eosinophil免疫突触。2)吞噬杯的形成。3)嗜酸性粒细胞巨噬细胞内在化。 b) Stepwise stages of macrophage-mediated eosinophil efferocytosis. 1, 2) Eosinophil undergoing apoptosis prior to 3) uptake and 4) engulfment by macrophages. c) Quantification of eosinophils engulfed by individual macrophages post-treatment with the indicated antibodies via live imaging (n=5). d) Left: quantification of eosinophil death (macrophage-engulfed and PO-PRO-1+嗜酸性粒细胞)通过实时成像,如总far-red-labelled嗜酸性粒细胞的百分比(n = 3),在巨噬细胞的存在;右:半胱天冬酶激活的变化评估通过流式细胞术使用CellEvent Caspase-3/7 (Casp3/7)试剂和表示为CD66b总额的百分比+Siglec-8+嗜酸性粒细胞(n = 3)。a、b)图像帧获得大约每5分钟,然后选择代表帧illlustration 15或30分钟的间隔。µm比例尺:2。c, d)数据意味着±扫描电镜。双向方差分析与图基和Šidak的多重比较。* *:p < 0.01;* * * *:p < 0.0001, benralizumab与父母fucosylated anti-interleukin-5受体α(PF anti-IL-5Rα)。介绍了统计值补充表S2。
进一步文档参与benralizumab-mediated吞噬作用和efferocytosis细胞事件,我们仔细检查了收购活细胞图像序列benralizumab治疗。当一个激活巨噬细胞和嗜酸性粒细胞是生活在接触,建立了吞噬突触(图3一1),其次是巨噬细胞的变化膜,形成伪足包围住嗜酸性粒细胞。网站的联系,一个吞噬杯(抑郁膜)形成(图3一2),启动嗜酸性粒细胞的吞没。30分钟内,膜末端封闭,创造新的时间(19,20.嗜酸性粒细胞退化(所需)图3一3和补充视频S6和S7)。除了住嗜酸性粒细胞的吞没,详细分析图像序列(补充视频S6和S7)意外发现嗜酸性粒细胞死前身体与巨噬细胞相互作用就是明证PO-PRO-1染料的公司(图3 b2和d,对面板)。识别的死后嗜酸性粒细胞,efferocytic突触形成(图3 b3),导致PO-PRO-1吞没和随后的退化+嗜酸性粒细胞通过激活巨噬细胞(图3 b4)。
benralizumab,个人巨噬细胞能吞噬四嗜酸性粒细胞与父母相比fucosylated控制(图3 c),导致吸收55%的嗜酸性粒细胞(图3 d(左)。在这些条件下,大约三分之二的嗜酸性粒细胞的吞噬作用(活的嗜酸性粒细胞),而三分之一接受efferocytosis(死去的嗜酸性粒细胞)。死CD66b+Siglec-8+嗜酸性粒细胞是由Caspase-3/7激活和PO-PRO-1染色(图3 d,对吧)。嗜酸性粒细胞的发生死亡之前,巨噬细胞参与建议额外的分泌机制的存在可能引发嗜酸性粒细胞死亡(17,18]。
NK细胞增强benralizumab-mediated嗜酸性粒细胞巨噬细胞死亡
当NK细胞被添加到混合细胞(巨噬细胞,1:5:15:嗜酸性粒细胞:NK细胞),我们观察到一个嗜酸性粒细胞吸收增强,生存或死亡,通过激活巨噬细胞,以应对benralizumab使用实时成像(图4一)。尽管激活巨噬细胞吞噬嗜酸性粒细胞总数保持不变的NK细胞(补充图S2),嗜酸性粒细胞吞噬了每个巨噬细胞的计数是大大增强,从四到八个嗜酸性粒细胞/巨噬细胞,通过添加benralizumab (图4一和补充视频S8)。量化的嗜酸性粒细胞死亡(所有PO-PRO-1吞没+住嗜酸性粒细胞)显示,死亡的比例增加了12%在NK细胞和嗜酸性粒细胞benralizumab (图4 b),导致67%的总嗜酸性粒细胞死亡(图4 b)。34.5% CD66b+Siglec-8+PO-PRO-1+嗜酸性粒细胞展出的相伴upregulation Caspase-3/7活动(图4 c和补充视频S9)。此外,图像分析强调了频繁,但瞬态,形成免疫突触的NK细胞和巨噬细胞之间,通常从9到14每巨噬细胞突触(补充视频S10和图4 d)。NK细胞观察不久参与激活巨噬细胞,然后分离而不被吞噬细胞。这些观察结果促使我们质疑NK-macrophage突触刺激影响巨噬细胞的行为。因此,我们描述benralizumab-mediated巨噬细胞激活和增强巨噬细胞吞噬NK细胞和细胞毒性反应。我们追踪细胞表面调理Fcγ受体调节我在CD163 (CD64)+巨噬细胞活化的细胞,作为一个指标在benralizumab-mediated嗜酸性粒细胞吞噬/ efferocytosis [21),并观察upregulation CD163增强CD64的表达式+巨噬细胞与benralizumab (图5一个和b)。CD64 upregulation伴随着增强表达的清道夫受体CD163(同形像的意思是荧光强度(MFI): 433;benralizumab MFI: 3998)和CD107a (LAMP-1),建议在巨噬细胞吞噬溶酶体降解实验的嗜酸性粒细胞(图5一个和b)。相比之下,抑制“唐't-eat-me”信号,信号调节蛋白α(SIRPα)[22激活巨噬细胞),降低(同形像MFI: 11 066;benralizumab MFI: 2497) (图5一个和b)。这些研究表明,benralizumab可以诱导巨噬细胞吞噬作用/ efferocytosis通过CD163 CD64和CD107a upregulation和伴随的,差别SIRPα对这些以及调节NK细胞介导巨噬细胞活化与其母相比fucosylated anti-IL-5Rα控制(图5一个和b)。

自然杀伤(NK)细胞增强benralizumab-induced嗜酸性粒细胞(Eos)消耗巨噬细胞(Mac)。人类巨噬细胞来源于外周血单核细胞后7天刺激与ng 50毫升−1巨噬细胞集落刺激因子。主要NK细胞和嗜酸性粒细胞隔绝自体健康的捐赠者,单独和每个细胞类型的标签。嗜酸性粒细胞与10 nM的预处理表示抗体,然后贴上了远红外的脂质膜染料和co-incubated green-labelled巨噬细胞和/或red-labelled NK细胞PO-PRO-1的存在。细胞混合6 h和流式细胞术或共焦显微镜分析活细胞成像。左):代表形象描绘增加巨噬细胞的吞噬活动(六个嗜酸性粒细胞的吞没)在NK细胞的存在benralizumab治疗(交互嗜酸性粒细胞划定的白色虚线);右:量化个人嗜酸性粒细胞吞噬的巨噬细胞与表示抗体通过后处理成像(n = 5)生活。µm比例尺:2。b)量化的嗜酸性粒细胞(macrophage-engulfed和PO-PRO-1死亡+嗜酸性粒细胞)通过实时成像,如总far-red-labelled嗜酸性粒细胞的百分比(n = 3),在巨噬细胞和NK细胞的存在。c)激活半胱天冬酶的变化评估通过流式细胞术使用CellEvent Caspase-3/7 (Casp3/7)试剂和表示为CD66b总额的百分比+Siglec-8+嗜酸性粒细胞(n = 3)。d)代表的形象重要免疫突触NK细胞和巨噬细胞之间。代表高度激活的巨噬细胞由绿色虚线。µm比例尺:2。两者数据意味着±扫描电镜。双向方差分析与图基的多重比较。*:p < 0.05;* * *:p < 0.001;* * * *:p < 0.0001, benralizumab与父母fucosylated anti-interleukin-5受体α(PF anti-IL-5Rα)。+:p < 0.05;+ + +:p < 0.001, Eos +苹果+ NK与Eos + Mac。介绍了统计值补充表S2。
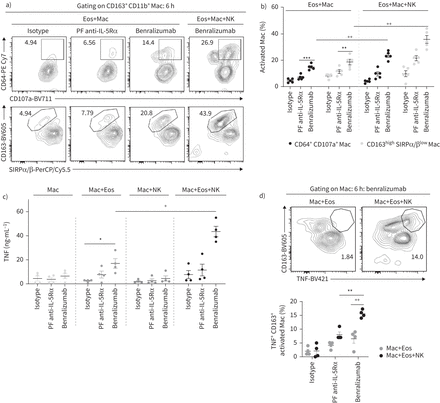
自然杀伤(NK)细胞增强benralizumab-mediated巨噬细胞(Mac)吞噬作用和细胞毒性。人类巨噬细胞来源于外周血单核细胞后7天刺激与ng 50毫升−1巨噬细胞集落刺激因子。主要NK细胞和嗜酸性粒细胞(Eos)与自体健康的捐赠者。嗜酸性粒细胞是预处理的10 nM的抗体。细胞混合6 h和流式细胞术分析。上层清液收集量化肿瘤坏死因子(TNF)的ELISA。a、b)巨噬细胞标志物的变化参与目标识别/掩饰(CD163 CD64和信号调节蛋白α/β(SIRPα/β))和吞噬溶酶体激活(CD107a),表示为CD163总额的百分比+CD11b+巨噬细胞(n = 5)。c) TNF含量量化由ELISA benralizumab后处理的存在和缺乏巨噬细胞和NK细胞(n = 4)。d)量化TNF的表达在巨噬细胞和NK细胞缺乏表示为CD163总额的百分比+CD11b+巨噬细胞(n = 4)。b, c, d)数据意味着±扫描电镜。双向方差分析测试图基的多重比较。*:p < 0.05;* *:p < 0.01;* * *:p < 0.001, benralizumab与控制(同形像或父fucosylated anti-interleukin-5受体α(PF anti-IL-5Rα))。+:p < 0.05;+ +:p < 0.01, Eos +苹果+ NK与Eos + Mac。介绍了统计值补充表S2。
macrophage-derived Benralizumab诱发TNFR1-mediated嗜酸性粒细胞凋亡的肿瘤坏死因子
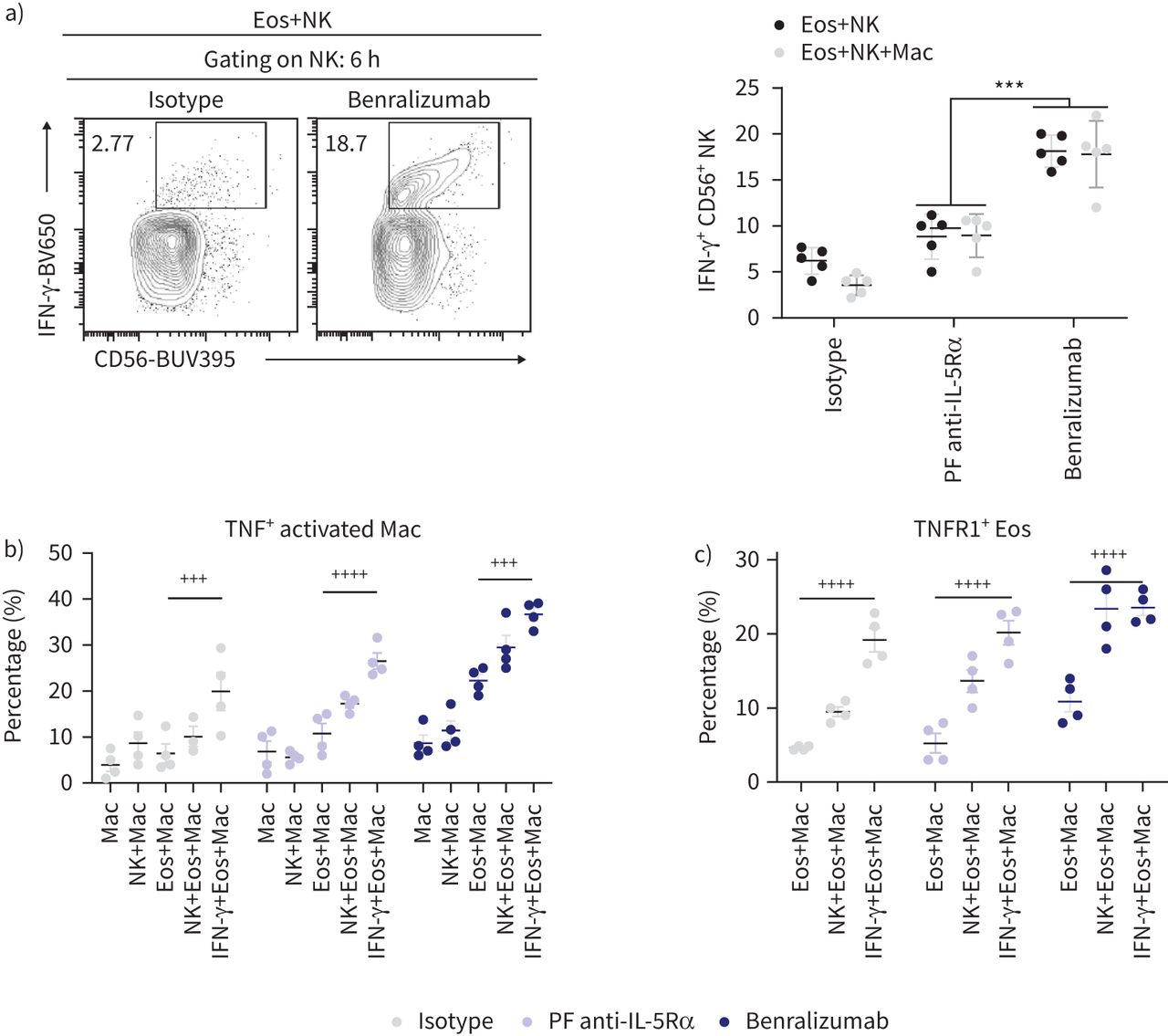
我们下一个调查benralizumab能否诱导macrophage-derived TNF的表达,促进TNFR1-mediated嗜酸性粒细胞凋亡。我们第一次评估人类巨噬细胞培养上清液中可溶性TNF独自或与benralizumab-treated嗜酸性粒细胞NK细胞的存在与否。虽然小但数量显著增加肿瘤坏死因子的上层清液中检测出与benralizumab-treated嗜酸性粒细胞巨噬细胞混合,一个健壮的upregulation被添加NK细胞诱导细胞混合。相比之下,TNF含量几乎可以在缺乏嗜酸性粒细胞(图5 c)。一直,我们已经展示了upregulation激活巨噬细胞(CD11b+CD163高)表达肿瘤坏死因子,其频率进一步增加在添加NK细胞(图5 d)。此外,我们发现了一个重要的upregulation TNFR1 CD66b+Siglec-8+嗜酸性粒细胞巨噬细胞培养,进一步提高通过添加混合NK细胞(图6)。综上所述,我们的数据突出目标细胞的要求生产benralizumab-induced TNF通过巨噬细胞TNFR1表达在嗜酸性粒细胞升高相关,也证实了鼓舞人心的NK细胞对肿瘤坏死因子和TNFR1表达的影响。验证是否benralizumab anti-eosinophilic活动相关的严重嗜酸性哮喘病人,我们证实TNFR1表达和IL-5Rα嗜酸性粒细胞,CD16 NK细胞和巨噬细胞,以及benralizumab-mediated嗜酸性粒细胞减少在体外(未发表的数据)。
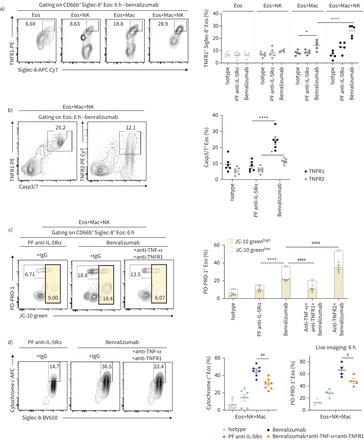
Benralizumab促进嗜酸性粒细胞(Eos)细胞凋亡的巨噬细胞(Mac)细胞毒性通过肿瘤坏死因子(TNF) /肿瘤坏死因子受体1 (TNFR1)通路。人类嗜酸性粒细胞与10 nM的表示预处理的抗体,然后贴上co-incubated巨噬细胞和自然杀伤(NK)细胞的存在PO-PRO-1 6 h。)量化TNFR1-induced表达式在嗜酸性粒细胞和巨噬细胞和NK细胞,表示为CD66b总额的百分比+Siglec-8+细胞(n = 4)。b) TNFR1表达与肿瘤坏死因子受体2 (TNFR2)凋亡Caspase-3/7+(Casp3/7)嗜酸性粒细胞巨噬细胞和NK细胞的存在和缺席,表示为CD66b总额的百分比+Siglec-8+细胞(n = 3)。c, d)凋亡的嗜酸性粒细胞减少,如评估c) PO-PRO-1 JC-10绿色线粒体膜电位测定和d)细胞色素c后,抑制TNF和TNFR1表示抗体的存在,巨噬细胞和NK细胞(n = 4)。数据均值±扫描电镜。双向方差分析与图基的多重比较。*:p < 0.05;* * * *:p < 0.0001, benralizumab与控制(同形像或父fucosylated anti-interleukin-5受体α(PF anti-IL-5Rα))。+ + + +:p < 0.0001, Eos +苹果+ NK与Eos + Mac。#:p < 0.05;# #:p < 0.01;# # # #:p < 0.0001,阻止抗体+ benralizumab与benralizumab。介绍了统计值补充表S2。
因为据报道,肿瘤坏死因子也绑定TNF受体2 (TNFR2)的激活与pro-survival通路在人类嗜酸性粒细胞(23),我们评估其表情嗜酸性粒细胞通过流式细胞术benralizumab治疗后6 h在巨噬细胞和NK细胞的存在。尽管大多数Caspase-3/7-expressing嗜酸性粒细胞TNFR1表达(25%),只有一半的人(12.5%)调节TNFR2表达式(图6 b),将caspase-dependent嗜酸性粒细胞死亡TNFR1 upregulation。尽管TNFR2表情凋亡嗜酸性粒细胞pro-survival信号显示激活,似乎赞成嗜酸性粒细胞凋亡频传。
防止肿瘤坏死因子信号通过TNFR2 (pro-survival),同时确保成功的封锁TNFR1-mediated嗜酸性粒细胞凋亡,anti-TNF-α和anti-TNFR1阻止抗体应用于细胞巨噬细胞、NK细胞和antibody-treated嗜酸性粒细胞。在这些条件下,早期凋亡的评估是通过线粒体膜电位的两极化,评估增加JC-10 CD66b绿色染色+Siglec-8+嗜酸性粒细胞。晚期凋亡被合并的增加表明PO-PRO-1染料在垂死的嗜酸性粒细胞和随之而来的减少JC-10绿色,扩散到其他细胞线粒体两极化的后车厢。正如预期的那样,我们观察到的早期细胞凋亡(JC-10绿色高和PO-PRO-1+嗜酸性粒细胞)以及细胞凋亡晚期(PO-PRO-1+和JC-10绿色低细胞)细胞接受benralizumab时(图6 c)。而阻塞独自TNFR2受损的嗜酸性粒细胞的生存(图6 c),抑制肿瘤坏死因子/ TNFR1导致部分但显著减少凋亡的嗜酸性粒细胞死亡就是明证PO-PRO-1和JC-10绿色的降低公司(图6 c),以上的差别以及对这些细胞色素c在CD66b+Siglec-8+嗜酸性粒细胞(图6 d左面板)。与我们的研究结果一致,活细胞成像分析证实了减少嗜酸性粒细胞凋亡在回应TNF / TNFR1封锁,在嗜酸性粒细胞减少PO-PRO-1标签(显示的图6 d右面板)。我们的数据表明巨噬细胞肿瘤坏死因子的存在/嗜酸性粒细胞TNFR1轴,当激活通过macrophage-benralizumab-eosinophil复合物的形成,导致嗜酸性粒细胞凋亡。
可以触发TNF-dependent巨噬细胞细胞毒性NK-derived IFN-γbenralizumab-stimulated嗜酸性粒细胞的存在
巨噬细胞激活是由几个NK-derived介质,包括最有效的激活细胞因子IFN-γ[14,17,24]。因此,我们量化细胞内IFN-γ表达NK细胞培养benralizumab-stimulated嗜酸性粒细胞。虽然IFN-γ表达CD56调节+NK细胞benralizumab-treated嗜酸性粒细胞的存在,其含量没有改变通过增加巨噬细胞的细胞结构,表明benralizumab-treated嗜酸性粒细胞发生上游NK细胞活化的巨噬细胞激活我们的实验条件下(图7)。
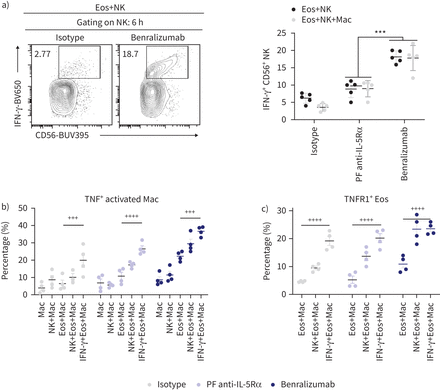
自然杀伤(NK)派生interferon-γ(IFN-γ)刺激肿瘤坏死因子(TNF)端依赖巨噬细胞细胞毒性benralizumab-stimulated嗜酸性粒细胞(Eos)的存在。)量化IFN-γ表达式在NK细胞benralizumab治疗,表示为CD56总额的百分比+CD94+细胞(n = 4)。b)量化TNF的表达在存在嗜酸性粒细胞巨噬细胞刺激后10 ng·毫升−1IFN-γ,表示为CD163总额的百分比+CD11b+巨噬细胞(n = 4)。c)量化的诱导表达TNFR1嗜酸性粒细胞巨噬细胞刺激后的10 ng·毫升−1IFN-γ,表示为CD66b总额的百分比+Siglec-8+细胞(n = 4)。数据均值±扫描电镜。双向方差分析与图基的多重比较。* * *:p < 0.001, benralizumab与父母fucosylated anti-interleukin-5受体α(PF anti-IL-5Rα)。+ + +:p < 0.001;+ + + +:p < 0.0001, 10 ng·毫升−1IFN-γ+ Eos + Mac与Eos + Mac。介绍了统计值补充表S2。
鉴于短期治疗IFN-γ可以增强ADCP的ADCC和巨噬细胞(17),我们调查的直接影响IFN-γbenralizumab-mediated巨噬细胞的嗜酸性粒细胞的反应和/或NK细胞。刺激激活CD163 IFN-γ诱导显著的肿瘤坏死因子表达高benralizumab-stimulated培养嗜酸性粒细胞巨噬细胞,比较与观察到的NK细胞(图7 b)。在这种情况下,TNFR1表达也是CD66b诱导+Siglec-8+嗜酸性粒细胞,达到类似水平NK细胞巨噬细胞培养(图7 c)。综上所述,我们的数据证实的刺激效果NK-derived IFN-γ上游macrophage-derived TNFR1-mediated嗜酸性粒细胞凋亡的肿瘤坏死因子。
最后,我们调查是否补体的激活也可能导致benralizumab-mediated嗜酸性粒细胞减少在体外。Benralizumab没有引入嗜酸性粒细胞的补体依赖细胞毒性,最有可能由于低拷贝数的表面IL-5Rα嗜酸性粒细胞(补充图S3)。
讨论
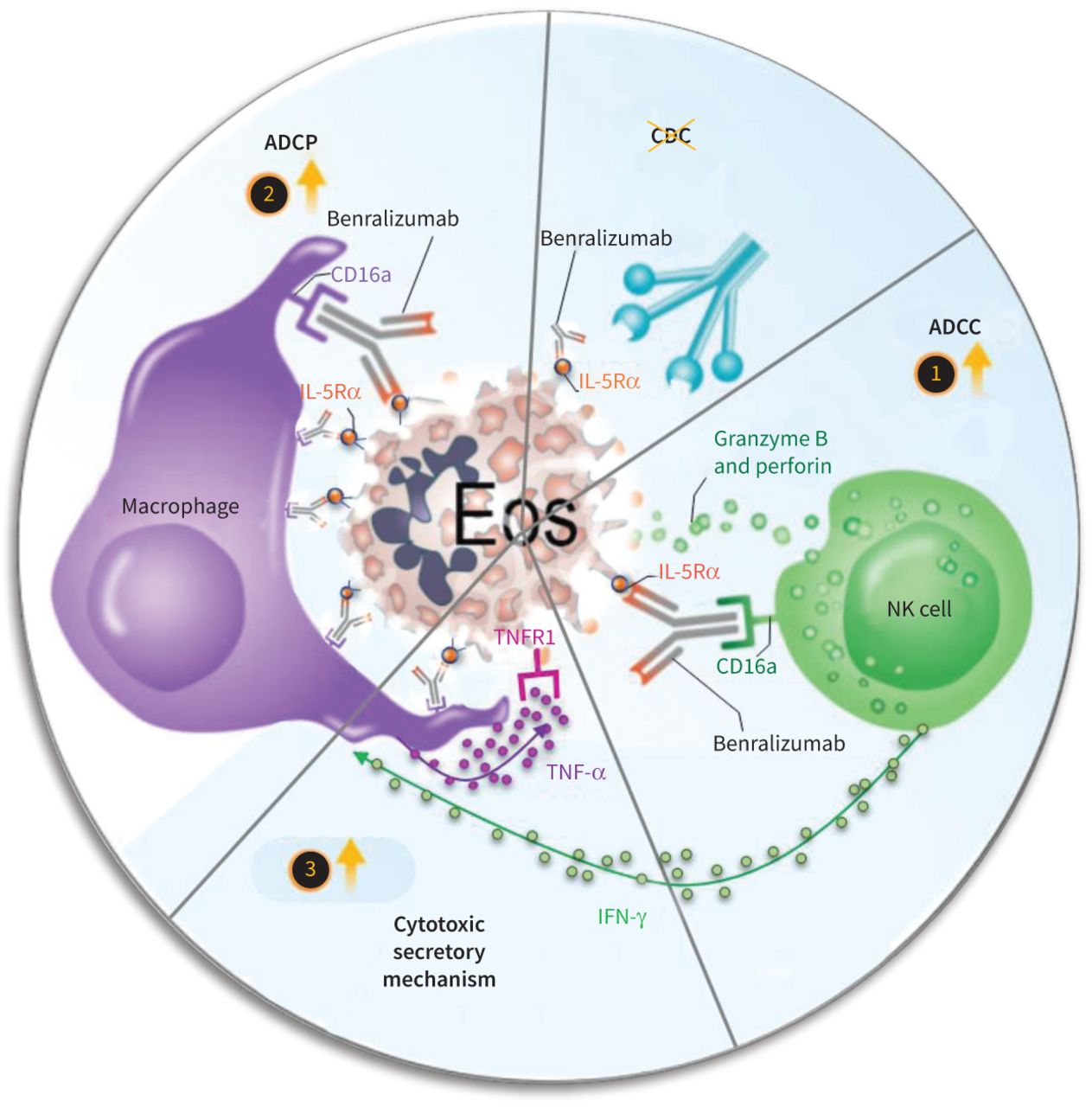
虽然嗜酸性粒细胞与宿主防御某些感染(25,26),他们的外围或组织水平升高与慢性炎性疾病,特别是严重的哮喘,嗜酸性肉芽肿病与polyangiitis (Churg-Strauss综合症),hypereosinophilic综合症和更多2,3,5]。嗜酸性粒细胞的复杂依赖IL-5生存和成熟鼓励发展的几种免疫疗法针对IL-5通路(4),包括IL-5Rα-targeting eosinophil-depleting抗体benralizumab [4,7,10]。阐明benralizumab的作用机理一直挑战由于其缺乏大鼠IL-5Rα和免疫球蛋白Fc受体生物学差异人类和老鼠。但是,通过使用相关的主要人类自体的细胞培养和real-time-lapse成像结合流式细胞术,我们发现了新的机制,可能导致benralizumab有力anti-eosinophilic活动在活的有机体内(图8)。我们第一次“视觉效果”的逐步过程部署benralizumab anti-eosinophilic活动也证明α-fucosylation benralizumab是所有这些活动的核心,是它的增强力量与其母相比fucosylated anti-IL-5Rα控制。
在NK-mediated ADCC化验,我们以前记录NK细胞毒性调停嗜酸性粒细胞benralizumab诱导细胞凋亡。在这里,我们进一步证实我们最初的发现与细胞色素的upregulationc在benralizumab-targeted嗜酸性粒细胞和半胱天冬酶的活动,以及相关的嗜酸性粒细胞形态变化,从一个嗜酸性粒细胞/ NK细胞突触的形成,最终导致嗜酸性粒细胞死亡。这种突触可能诱发NK细胞毒性,刺激信号可以绕过抑制剂对NK细胞受体表达的作用,即杀手immunoglobulin-like受体(吉珥)[27]。这样的机制可以解释的效力benralizumab-mediated嗜酸性粒细胞减少观察反应良好,尽管增加吉珥表情NK细胞严重嗜酸性哮喘受试者(28]。
此外,我们的数据突出新机制,死亡benralizumab能引起嗜酸性粒细胞巨噬细胞通过两个不同的过程涉及的嗜酸性粒细胞巨噬细胞吞噬作用的直接吸收/ efferocytosis (ADCP)或TNFR1-mediated嗜酸性粒细胞凋亡的诱导通过macrophage-derived TNF。后者被进一步增强NK细胞衍生IFN-γ。尽管TNF / TNFR1此前报道提高嗜酸性粒细胞生存通过NF-κB激活(11,29日]通过物/ AP-1通路(30.),这个途径抑制了向全世界揭示肿瘤坏死因子诱导嗜酸性粒细胞凋亡的能力(31日]。在这里,我们表明,增加TNFR1表达与upregulation几个凋亡细胞死亡的标志,尤其是细胞色素c、膜两极化和Caspase-3/7活动在我们的条件下,因此主张的直接贡献TNFR1嗜酸性粒细胞凋亡,看似通过NF-kB pro-apoptotic通路的抑制或激活。验证是否benralizumab anti-eosinophilic活动相关的严重嗜酸性哮喘病人,我们证实TNFR1表达和IL-5Rα嗜酸性粒细胞,CD16 NK细胞和巨噬细胞,以及benralizumab-mediated嗜酸性粒细胞减少在体外(未发表的数据)。与高浓度的granzyme B和IFN-γ,一个完整的嗜酸性粒细胞损耗由benralizumab prednisone-dependent严重哮喘患者痰中演示(28,32];这个主张上述效应机制的参与严重哮喘患者的气道。
虽然肿瘤坏死因子被认为是营养因素推迟嗜酸性粒细胞凋亡在哮喘29日,33),在这里,我们表明,诱导表达的肿瘤坏死因子在免疫突触,由benralizumab巨噬细胞和嗜酸性粒细胞之间增强TNFR1-mediated嗜酸性粒细胞pro-apoptotic信号在体外。而肿瘤坏死因子抑制剂中免疫疗法,如golimumab没能证明有利的风险在严重的哮喘和保留一个小群(表现出有限的效果34],benralizumab似乎部署几个耗尽嗜酸性粒细胞严重哮喘的机制,包括新发现的肿瘤坏死因子/ TNFR1-mediated嗜酸性粒细胞凋亡通过benralizumab-directed免疫突触,从而延长其有效anti-eosinophilic活动在诊所。进一步调查才能阐明肿瘤坏死因子/ TNFR1的下游信号通路促进嗜酸性粒细胞凋亡benralizumab治疗的反应。此外,ADCP的嗜酸性粒细胞巨噬细胞可能代表一个重要机制,从组织嗜酸性粒细胞的“沉默”删除,就是明证在体外通过实时成像(补充视频S6-S10)。我们的研究结果与临床观察一致,benralizumab-mediated嗜酸性粒细胞枯竭之际,减少了免费的嗜酸性颗粒,如eosinophil-derived神经毒素和嗜酸性粒细胞阳离子蛋白,在严重哮喘患者的血清35]。然而,在最近的一项研究在hypereosinophilic综合症患者,K独角仙等。(36)描述的症状包括发烧、发冷、头痛、恶心和疲劳,开始∼6 h后第一个benralizumab的剂量。这些自限性的情节没有复发与随后的剂量。有趣的是,预处理的嗜酸性粒细胞数量和NK细胞和NK细胞的活化状态与治疗反应的发展,表明组织本地化和其他效应机制的参与,如本文中所描述的,可能控制这种瞬态反应患者的外观。
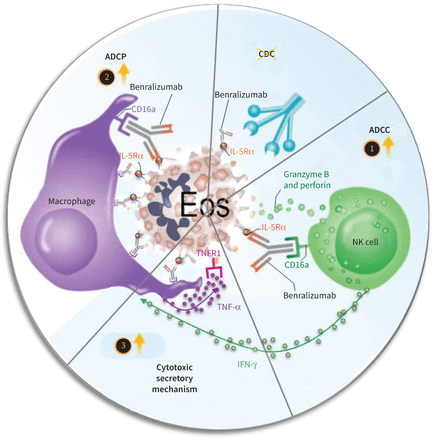
拟议机制benralizumab-induced嗜酸性粒细胞(Eos)损耗通过锁定细胞介导细胞毒性(ADCC),锁定细胞吞噬作用(ADCP)和肿瘤坏死factor-α(TNF-α)端依赖巨噬细胞细胞毒性。1)ADCC过程,benralizumab嗜酸性粒细胞凋亡诱导激活自然杀伤(NK)细胞。2)另外,ADCP通过巨噬细胞耗尽benralizumab促进嗜酸性粒细胞通过吞噬作用或efferocytosis。3)激活NK细胞释放interferon-γ等刺激因素(IFN-γ)诱导巨噬细胞细胞毒性通过TNF-α可以触发肿瘤坏死因子受体1 (TNFR1)端依赖嗜酸性粒细胞凋亡;这些凋亡细胞被巨噬细胞efferocytosis移除。最后,补体依赖细胞毒性(CDC)似乎不参与benralizumab-mediated嗜酸性粒细胞减少。IL-5Rα:interleukin-5受体α。
在多大程度上benralizumab部署描述机制(图8)患者组织仍然未知,最有可能依赖于炎症的程度,类型的组织和效应细胞的存在。除了嗜酸性粒细胞,最近的一篇论文报道benralizumab-mediated损耗的新目标细胞,即IL-5Rα-expressing 2型先天淋巴细胞,IL-5[的主要来源28]。其他研究发现存在自身免疫复合物在不受控制的严重哮喘受试者,可能导致气道炎性环境导致增强IFN-γ和TNF水平(37,38),从而允许我们推测autoimmune-dependent机制以及感染可能促进benralizumab-mediated嗜酸性粒细胞损耗严重嗜酸性哮喘。
我们不能排除这种可能性,附加效应细胞表达CD16,如中性粒细胞和树突细胞,可能导致benralizumab anti-eosinophilic活动组织(39,40]。同时,遗传多态性的基因编码CD16可能调整其活动(41]。这些问题需要进一步调查。因为afucosylated单克隆抗体,比如benralizumab,代表一个新兴阶级的疗法(42),了解他们的效应函数高度相关benralizumab本身之外,对其他cell-depleting抗体在诊所。
补充材料
补充材料
请注意:补充材料并不是由编辑部,编辑和上传已由作者提供。
补充视频S1。相关视频S1、S2和S3图1。实时的视频benralizumab诱导caspase-dependent通过ADCC嗜酸性粒细胞凋亡。主要人类NK细胞和嗜酸性粒细胞是从健康的捐赠者和green-labelled单独孤立的。NK细胞与绿色LysoTracker加载。生活形象benralizumab-mediated ADCC, red-labelled嗜酸性粒细胞是使用1μg·毫升−1的父母fucosylated anti-IL-5Rα(视频S1)或benralizumab(视频S2),然后与green-labelled NK细胞混合PO-PRO-1 4 h。后来,混合细胞和共焦显微镜成像住2 h。图像获得每2或5分钟。erj - 04306 - 2020. - video_s1
补充视频S2。erj - 04306 - 2020. - video_s2
补充视频S3。实时视频illustratingcaspase激活。CellEvent Caspase-3/7试剂添加之前活细胞采集。怀特Caspase-positive细胞中描述。erj - 04306 - 2020. - video_s3
补充视频S4。相关视频S4和S5图2。实时的视频benralizumab-mediatied嗜酸性粒细胞ADCC NK细胞。视频S4强调了生物检查站在评估NK细胞(绿色)嗜酸性粒细胞(红色),从免疫突触,NK-green颗粒释放到red-esosinophilic胞质嗜伊红血球膜起泡、损失的细胞完整性,也描述了视频S5。Green-labelled NK细胞溶素的颗粒聚集在免疫突触和停靠red-labelled嗜酸性粒细胞的质膜进行了细胞凋亡的团结就是明证PO-PRO-1蓝核染料。补充视频S4。erj - 04306 - 2020. - video_s4
补充视频S5。erj - 04306 - 2020. - video_s5
补充视频S6。视频S6和S7有关图3。实时的视频benralizumab-mediated通过ADCP嗜酸性粒细胞凋亡。生活形象benralizumab-mediated ADCP(视频S6和S7),主要单核细胞分化成巨噬细胞为6天。然后,嗜酸性粒细胞被孤立于自体健康的捐赠者和单独的标签。预处理后1μg·毫升−1benralizumab far-red-labelled嗜酸性粒细胞(白色)核染料PO-PRO-1与green-labelled巨噬细胞和蓝色的混合。后来,混合细胞活6 h和共焦显微镜的成像。图片是每5分钟。erj - 04306 - 2020. - video_s6
S7补充视频。erj - 04306 - 2020. - video_s7
S8补充视频。视频S8, S9、S10有关图4。实时视频强调增强NK benralizumab-mediated巨噬细胞吞噬作用/ efferocytosis和NK-macrophage免疫突触。实时视频S8说明了NK(红色)对巨噬细胞的影响(绿色)吞噬benralizumab引起的行为。erj - 04306 - 2020. - video_s8
补充视频S9。实时视频S9表明NK细胞和巨噬细胞吞噬积极互动(绿色)在benralizumab治疗。嗜酸性粒细胞的标签在核染料PO-PRO-1白色和蓝色的。erj - 04306 - 2020. - video_s9
S10补充视频。实时视频S10表明白标签嗜酸性粒细胞加入蓝色染料,apoposis的一个特点,是通过积极green-labeled巨噬细胞(efferocytosis)也能吞没另一个red-labelled NK细胞。erj - 04306 - 2020. - video_s10
可共享的PDF
确认
我们感谢理查德·n·汉纳(美国马里兰州盖瑟斯堡,阿斯利康,)他的援助在成像和约翰·蒂(美国阿斯利康,马里兰州)审查统计测试。
脚注
作者的贡献:r . Dagher协调项目,导致了研究的概念,建立了研究设计,负责数据采集/分析/解释和起草了手稿。诉Kumar执行细胞因子测量/分析、流式细胞仪采集/分析TNFR1-mediated细胞凋亡,并帮助起草了手稿。点Copenhaver执行半胱天冬酶试验和流式细胞仪采集/分析ADCC过程。m . Ghaedi提供科学支持。j·博伊德帮助获得生活ADCC和ADCP过程图像。加拉格尔疾控中心进行分析。p·纽伯尔德和嗜11为本研究提供了指导。r . Kolbeck研究概念和监督,负责获取资金,并起草手稿。
利益冲突:r . Dagher受雇于和阿斯利康的股东。
利益冲突:诉库马尔是受雇于和阿斯利康的股东。
利益冲突:上午Copenhaver是阿斯利康的前雇员和股东,并受雇于和武田制药的股东。
利益冲突:美国加拉格尔是阿斯利康的前雇员。
利益冲突:m . Ghaedi受雇于和阿斯利康的股东。
利益冲突:j·博伊德是受雇于和阿斯利康的股东。
利益冲突:p·纽伯尔德是受雇于和阿斯利康的股东。
利益冲突:嗜教训了阿斯利康股份的所有者。
利益冲突:r . Kolbeck是阿斯利康的所有者的股票。
支持声明:这项工作完全由阿斯利康。
- 收到了2020年11月25日。
- 接受2021年7月7日。
- 版权©2022年作者。
这个版本分布在创作共用署名非商业性许可证的条款4.0。商业生殖权利和权限接触权限在}{ersnet.org











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}