





文摘
模型小鼠肺癌的持续进步而导致不仅新理解人类肺癌的分子通路管理的手段,但它也创造了一个巨大的水库的替代工具来测试治疗对恶性肿瘤。更复杂的躯体nonsmall细胞肺癌小鼠模型,小细胞肺癌和肺鳞状细胞癌已经生成密切模仿人类肺癌。这些模型使我们能够确定起源的细胞和干细胞的作用在维护的各种类型的肺癌。此外,肺癌干预研究的结果现在开始揭示这些体细胞的潜能作为强大的临床前模型小鼠模型。
“肺癌”系列
Brambilla编辑c .
在本系列11号
全基因组检测方法进展基因改变在人类肺癌导致越来越多的肺癌症相关基因的鉴定。全基因组关联研究,不管他们是基于单核苷酸多态性阵列研究或检测基因拷贝数的变化通过比较基因组杂交数组,链接肺癌症相关基因突变的发生和频率的定义明确的表型大量人类肺癌。这些肺癌症相关基因提供了巨大的潜力为肺癌介入治疗靶点。目标验证然后发生在体外干预研究的这些特定的基因突变和各自的分子途径在人类肺癌细胞系。然而,在体外细胞培养的研究是有限的,不能完全模拟更加复杂在活的有机体内出现肿瘤发生和应对肿瘤治疗。发展中肺癌小鼠模型港口特定突变无疑将提供一个进一步更好的洞察突变特异对肺肿瘤生物学的影响。此外,高度的pathophysological相似性肺肿瘤小鼠模型和人类同行将有可能为临床前测试使用这些小鼠模型。各种干预策略针对特定突变可以测试基于更好的特异性和有效性评价小鼠肺肿瘤的每一个发展阶段。不断创新的技术来操纵鼠标基因组使我们调整复合肺癌小鼠模型以这样一种方式,他们开始繁殖更复杂的人类肺癌在一个更高的学位。互补,转基因小鼠模型的数量对肺癌是不断扩大。
首先对肺癌小鼠模型
使用小鼠模型自发或化学诱导肺肿瘤有着悠久的历史。而易感性和自发性肺肿瘤发病率的mouse-inbred菌株之间的不同,他们所有的肺癌分子病理与人类有很多相似之处1。这显然是建立在早期的研究中定义化学致癌物质被用来诱导肺肿瘤1。自发和诱发肺肿瘤的发生率最高(61%)的都是敏感菌株如J和驻波,但非常低(6%)在高度耐药菌株C57BL6和DBA在2岁男性1。与人类肺癌有着复杂的分子遗传学和四个不同肿瘤类型容易metastasise,自发和化学诱导小鼠的肺部病变常导致肺腺瘤2几乎从不metastasise更罕见的腺癌。小鼠肺肿瘤发展显示了初始增生性焦点在细支气管和肺泡,最终成为良性腺瘤和腺癌2。最可再生的肿瘤明显延迟取决于应变敏感性carcinogen-induction协议和/或应用程序。最强有力的致癌物质是吸烟致癌物质,如多环芳烃、烟草亚硝胺和苯并[a]芘(B(一)P)3。然而,吸烟本身就足以引起反复在休战后/ J小鼠肺肿瘤曝光时间其次是一个至关重要的4个月的恢复期4。接触B6C3F1雌性老鼠的一生(30个月)吸烟导致48%肺良性和恶性肿瘤通过不同(epi)基因通路5。因此,有明显的差异(自发)小鼠和人肺肿瘤。尤其难复制特征明显3人类呼吸道上皮病变6。然而,主要的组织病理学相似之处仍然和分子自发的描述和carcinogen-induced小鼠肺肿瘤显示高度的遗传损伤人类同行相比7。著名的早期事件激活Kras突变的发生7在增生性病变。除了表达cMYC,著名的肿瘤抑制基因失活,如肿瘤抑制蛋白53 (Trp53),脆性组氨酸三蛋白质(fhit),腺瘤息肉病杆菌蛋白质(APC),视网膜母细胞瘤(RB)的蛋白质,产生了突变蛋白在结直肠癌患者p16 (MCC)INK4A(CDKN2A)7,容易发生;最常见的腺瘤只有零星的进步发展为腺癌。对肺癌易感性的差异发展各种小鼠压力依然存在,然而,非常有趣。最敏感菌株,如/ J和BALB / C,在喀斯特的第2内含子多态性8,9和一个CDKN2a多态性被发现在BALB / C,影响他们对肺癌的敏感性。由于全基因组序列分析,努力更有趣的各种老鼠菌株可能遵循的多态性信息,可以与人类同源的同行中发现大型面板的人类肺癌基因组数据。此外,正如我们将看到在本文之后,更直接的转基因模型将会非常适合进入耐药菌株为了方便直接搜索修改肺肿瘤易感性的基因。
转基因小鼠
第一代
第一代转基因模型是基于异位转基因表达不同的启动子的控制下。表达主要是针对特定子集的肺上皮细胞:表面活性剂蛋白质c子指导主要表达II型肺泡细胞而克拉拉细胞secreatory蛋白质(CCSP) / CC10发起人主要目标nonciliated分泌细胞(Clara)航空公司。SV40标签(猴病毒大t抗原)持续表达CCSP后面10,11或表面活性剂蛋白(SP) - c推动者12。尽管每个肿瘤起源于克拉拉或II型肺泡细胞,它们都没有转移导致积极的腺癌非常相似13。一个类似的策略是用于不同的致癌基因(如cRaf和cMyc14),然而,温和的表型,最终作为主要转基因老鼠在腺瘤和一些进展腺癌,又没有任何转移。
许多著名的遗传病变发现在人类肺癌显然链接著名的肿瘤抑制基因的失活15肺癌的发展。最初试图模仿这些病变与肺癌的传统基因敲除小鼠的研究相当有限的成功对肺癌的发病。细胞缺失的主要原因是许多重要的肿瘤抑制基因(如视网膜母细胞瘤基因(Rb)和霍奇金病tumour-1同族体基因(Wt-1)16)导致胚或围产期杀伤力。不必要的肿瘤抑制基因敲除小鼠有更长的寿命通常有一个非常广泛的肿瘤谱的肺肿瘤形成的比例较小。因此,Trp53,p16INK4A和p19东盟地区论坛17零等位基因纯合子小鼠很少患肺腺癌。然而,类似的突变引入内生Trp53等位基因,如突出Li-Fraumeni患者发现,生成Trp53R270H / +和Trp53R172H / +有不同的肿瘤谱相比,哪一个Trp53+ / -18,尽管他们的平均生存时间是相同的。有趣的是这些老鼠,特别是Trp53R270H / +和Trp53R270H / -导致了更多的恶性肺腺癌,粘连形成,甚至转移这永远不会发生Trp53- / -老鼠。这些结果表明,“人性化”Trp53突变产生更大的影响比完整的肺肿瘤进展Trp53损失18,19。目标基因在人类肺癌肿瘤发生早期删除,如完整的集群在染色体3 . 3,表明,杂合的删除这个370 kb地区肺癌显示没有明显的倾向发展虽然纯合子缺失导致胚的杀伤力20.。一个更具体的候选肿瘤抑制基因的缺失3号染色体上RassF1a基因,FHIT基因和VHL显示,31%的RassF1a基因- / -小鼠自发的产生主要是淋巴瘤也肺腺瘤21。治疗RassF1a基因- / -小鼠B[一]P或聚氨酯导致了更高的肺肿瘤。没有观察到自发的肺肿瘤Fhit基因- / -或Vhl+ / -老鼠,但44%的Fhit基因- / -/ Vhl+ / -老鼠发达腺癌按年龄2岁。再次使用诱变剂,如二甲基亚硝胺导致100%腺瘤和腺癌感应Fhit基因- / -/ Vhl+ / -老鼠和甚至在40%的腺瘤Fhit基因- / -老鼠按年龄20个月22。这表明这些基因敲除小鼠的作用概括一个模式类似于人类早期肺癌的发展模式。
一种不同的方法来解决肺癌发病是激活癌基因敲入等位基因的使用。这方面的一个例子是基于体细胞喀斯特激活通过一个致癌喀斯特G12D敲入等位基因(喀斯特水为),这表示只有在一个自发重组事件(图。1⇓)23。通过这种方式,零星的喀斯特G12D表达发生在一个内生的水平,进而增强肺腺癌的有效发展。然而,这些老鼠还会表现出喀斯特的其他肿瘤病变G12D表达式是不限于肺上皮组织。
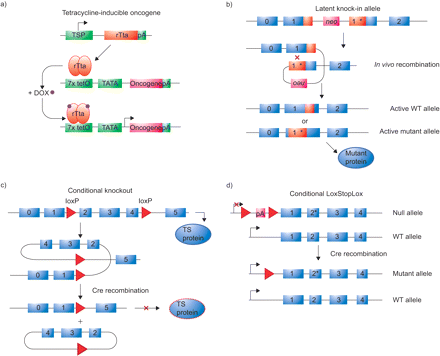
概述系统在大多数转基因小鼠模型用于肺癌。一)tetracycline-inducible bitransgenic系统。修复启动子(TSP)驱动器的表达Tet-inducible反式激活因子(rtTA)。在强力霉素(阿霉素),rtTA二聚体可以绑定到七tetO(7 x tetO)序列和转录激活癌基因的最小塔塔启动子。b)的激活等位基因确保随机致癌基因的激活通过自发的在活的有机体内沉默突变等位基因的重组。新霉素抗性(neo)基因分离野生型(WT),否则相同的外显子的突变复制。重组后,只有WT等位基因或等位基因的激活突变是产生致癌作用。c)有条件的基因失活是基于Cre / loxP recombination-mediated删除一个或多个外显子。特定场地Cre重组酶诱发突变通过重组两个直接面向LoxP序列。因此,控制Cre重组酶活动确保最终的肿瘤抑制基因失活(TS)成各种组织的选择。d)相反,条件LoxStopLox等位基因利用强大的转录停止,两侧是两个lox网站,在编码外显子的面前。由此产生的无效等位基因可能成为致癌突变等位基因的转录后停止元素通过Cre-mediated重组。pA: polyadenosine。
第二代
为了完善目前的小鼠模型,更好的方法复制真实的致癌基因的表达模式在肺肿瘤发生必须考虑。此外,一般敲入或敲除过程只差代表遗传事件发生在零星的肺癌。太大的一个扩展的主要致癌基因的表达和/或肿瘤抑制基因的失活创建一个微环境,根本不对应于零星的癌症发展只是一个有限的肿瘤细胞与正常细胞包围和互动24。条件调节致癌基因的时空表达或删除肿瘤抑制基因的体细胞组织的选择可以更准确地模拟在活的有机体内情况导致零星的癌症发病(图。1⇑)。这就是为什么第二代的肺癌小鼠模型利用一个条件bitransgenic四环素诱导系统25。多数情况下,反向tetracycline-controlled反式激活因子(rtTA)诱导系统使用。第一个转基因rtTA元素背后的组织发起人保证rtTA表达式一个细胞或组织类型的选择。这然后加上第二个转基因,转基因组成的目标tetracycline-responsive背后基因启动子(TetO7)。四环素、强力霉素的存在保证了稳定rtTA元素的交互TetO7目标基因启动子,进而表达。
因此,开/关目标基因表达可能取决于政府或撤回四环素/强力霉素26。这两个SP-C-rtTA和CCSP-rtTA转基因27已被用于指挥强力霉素敏感rtTA肺泡II型或克拉拉细胞。几个bitransgenic老鼠等CCSP-rtTA; TetO7FGF-728和CCSP-rtTA; TetO7喀斯特G12D29日已经成功地用于诱导肺损伤。诱导FGF-7引起最初的上皮细胞增生腺瘤后增生强力霉素应用紧随其后。所有增生撤回强力霉素后消失28。然而,喀斯特G12D感应引起上皮细胞增生、腺瘤的增生和强力霉素应用2个月后,多个腺瘤和腺癌。再次,没有病变可以检测到1个月后强力霉素撤军29日。当CCSP-rtTA; TetO7喀斯特G12D等位基因是与传统相结合Trp53或Ink4a /论坛无效等位基因,腺癌与恶性表型已经1个月后出现强力霉素的应用程序,因此显示mutant-Kras的协同作用和肿瘤抑制基因缺陷。然而,即使在这些化合物Tet-inducible小鼠模型,所有病变强力霉素撤军后消失。这表明mutant-Kras作为“驾驶”致癌基因的重要性不仅在肿瘤发病,而且在维护腺癌在这些小鼠模型。虽然与人类腺癌组织病理学相似之处非常明显,未发现转移从这些小鼠腺癌出现29日。
模拟更复杂的肺肿瘤基因在人类肺癌需要扩展方法与其他条件遗传系统。Flp Cre / loxP或/ FRT系统24,30.提供优秀的工具,通过引入有限数量的细胞分化的体细胞突变的选择,其他细胞的发育完全的肺保持正常23,31日。简而言之,目标区域的突变,两侧loxP(也称为“液氧”)或FRT序列网站,介绍了通过删除各自特定站点可Cre或隔爆。因此,在肿瘤抑制基因的情况下,条件hypomorphic突变或零等位基因,有几个(非)编码外显子液氧和,因此,被删除的相应的重组酶(图。1⇑)。相反,液氧转录停止(Lox-Stop-Lox或LSL)的致癌基因或敲入等位基因可以控制各自的条件激活32。这个条件的决定因素的控制方法是时空Cre或FRT重组酶表达。为此,几个Cre转基因线已经生成,有或没有春节诱导启动子27。除此之外,Cre-mediated重组也可以通过政府的工程Adeno-Cre病毒通过鼻腔和气管滴注法31日,33。后一种方法的一个优势是,有限的成人肺细胞可以有针对性的在一个非常简洁,本地化和及时。该方法的有效性进行了测试条件的等位基因喀斯特G12D和喀斯特G12V31日,33,34。感染成人肺Adeno-Cre病毒迅速导致腺瘤肺泡增生的发病,紧随其后的是最后发展腺瘤和腺癌在感染后3 - 4个月。虽然8个月的延迟也观察到34能找到,没有转移的模型。很可能一个喀斯特激活并不足以让腺癌进展到一个更高的恶性肿瘤作为完全转移病灶的需要。然而,这些简单的实验揭示喀斯特的重要作用在人类肺癌发病和进展17,34。这个模型的另一个重要方面是,肺肿瘤多样性可以控制的剂量的Adeno-Cre病毒只感染肺上皮细胞的一个子集。加之受控Adeno-Cre应用程序的时间点,确实保证精确的模仿人类肺癌的零星的性格发展。
然而,人们必须小心注意,可变性Adeno-Cre病毒的交付和感染(特别是鼻内方法)可能会导致不一致的实验结果。不过这个多才多艺的方法仍然是强大的,它像人类肺癌事件。
在未来我们审查的一部分我们要给一个更新最近的进展对人类肺癌小鼠模型。这我们将把重点放在程序的三个主要类型的肺癌,即nonsmall细胞肺癌(NSCLC),小细胞肺癌(SCLC)和鳞状细胞癌(SCC)。
可用转基因小鼠模型的类型、因果genotype-phenotypic转化研究的相关性和影响将简要地讨论了这些主要的肺癌类型。
模型对非小细胞肺癌
到目前为止,大多数小鼠模型生成肺腺癌与恶性肿瘤的不同阶段。正如我们已经讨论的,开车肺肿瘤发生的一个常用方法是通过条件或自发的激活喀斯特突变17。然而,很明显,我们需要进一步理解的更复杂的小鼠模型的高度复杂的肺癌基因(图。2⇓)。这些小鼠模型的验证需要的知识如何密切肺癌小鼠模型类似于人类。几项研究使用跨物种之间的基因表达谱比较小鼠和人肺肿瘤癌症信号通路中找出相似点35,36。从Kras基因表达数据的集成水为小鼠模型和KRAS突变的人类肺肿瘤的确显示出明显的重叠但也透露KRAS突变在人类肺癌的基因表达特征本身35。这些结果提出了强大的分子标准评估老鼠和人类肺癌之间的相似性,以及新的可能性,进一步阐明重要基因控制少特征明显肺癌通路。自激活喀斯特突变模型概括一些人类肺肿瘤表型相当好,仔细分析早期肺肿瘤进行了初始的事件37,38。两者的结合CCSPCC10-Cre重组酶,LSL喀斯特G12D等位基因37导致进步的表型的细胞异型性,最后腺瘤和腺癌。这些病变是专门从支气管上皮细胞,但伴随着强烈的炎症反应通过各种趋化因子的表达。喀斯特激活能,因此,作为一个优秀的模型来研究复杂的转换肺上皮细胞和炎症细胞之间的相互作用,趋化因子和早期肿瘤的发展。另一个模型生成,良性肺肿瘤病变利用bitransgenic Tet-inducible喀斯特G12C等位基因可以表达在克拉拉和/或肺泡II型细胞27,38。只有轻度非典型增生或良性腺瘤被发现后,甚至诱导喀斯特G12C不再表达≥12个月,肿瘤进展可以被探测到。抑制喀斯特G12C表达式通过强力霉素提取1月导致一个完整的任何早期的增生性病变的逆转。因此,这两个喀斯特G12C和喀斯特G12D致癌基因需要维护一个肿瘤表型。肿瘤低外显率的一种解释喀斯特G12C等位基因可能是缺乏一种蛋白激酶和物信号通路的激活。后者在强烈的对比观察前面描述的影响喀斯特G12D模型。
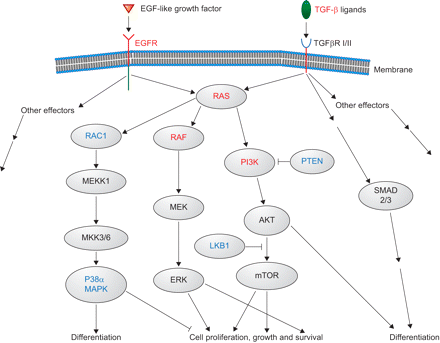
原理的概述的主要信号通路在大多数nonsmall受损细胞肺癌模型。激活信号网络的发生通过致癌基因的激活突变(红色所示)或灭活肿瘤抑制基因(蓝色)所示。信号级联的核心是RAS(鼠肉瘤病毒致癌基因同系物)及其刺激RAF-MEK-ERK(丝氨酸/苏氨酸激酶raf-mitogen-activated蛋白激酶kinase-extracellular signal-regulated激酶激酶)和PI3K(磷脂酰肌醇3-kinase)通路。激活表皮生长因子受体(EGFR)的突变的主要模型作为受体酪氨酸激酶上游的RAS。至于转化生长因子(TGF) -β,人们必须承认其双重角色作为一个肿瘤抑制基因在早期阶段的肺肿瘤发生和pro-oncogenic因素更高级和肺转移性肿瘤。注意,RAC1 (Ras-related C3肉毒杆菌毒素基质1,小GTPase)产生致癌活性与RAS合作,自灭活RAC1模型显示RAS-driven肺肿瘤发生的一个障碍。EGF:表皮生长因子;TGFβR: TGF-β受体;Mekk1:有丝分裂原活化蛋白激酶激酶激酶1或MAP3K1丝氨酸苏氨酸激酶;Mkk3/6:增殖蛋白激酶激酶3或MAP2K3,丝氨酸苏氨酸激酶; p38α-MAPK: mitogen activated protein kinase p38α or MAPK14; PTEN: phosphatase and tensin homolog; AKT: serine/threonine kinase also known as protein kinase B; LKB1: serine/threonine kianse LKB1 also known as serine/threonine protein kinase 11, STK11: mTOR: mammalian target of rapamycin, serine/threonine protein kinase; SMAD 2/3: TGF-β-dependent gene transcription factors.
这相当令人费解喀斯特但不是极品或国家管制当局方面在肺癌癌突变主要是发现39。一个合理的解释可以是,Ras蛋白亚型的不同呈现不同的癌蛋白活动,或各自的差异基因调控元素赋予组织表达突变拉基因在其相应的肿瘤。此外,必须注意喀斯特基因编码两个亚型,4 a和4 b, C末端的差别。主要的C末端喀斯特4 b同种型倾向于排斥的转录后修饰,如farnesylation或geranylgeranylation40。然而,4喀斯特翻译修饰,包括棕榈酰基共价连接,hra和国家管制当局方面。后者修改给国家管制当局方面,hra和喀斯特4一个独特的信号活动通过促进他们在特定的本地化microdomains膜41。优雅的最近的一项研究阐发Kras突变在肺癌特异性交换hra或喀斯特4 b cDNA到原始的喀斯特基因通过生成极品(hraKI)或喀斯特4 b(喀斯特KI)敲入小鼠。令人惊讶的是,urethane-induced肺肿瘤的数量极品KI /吻相比要高得多喀斯特KI /吻老鼠,后者产生更少比野生型小鼠(WT)控制肿瘤42。有趣的是,当突变进行分析喀斯特KI / wt杂合的老鼠,他们只检测到表达4喀斯特的WT等位基因,以及喀斯特4 b。这导致了这样的结论:只有一个突变喀斯特4等位基因,毕竟,与hra共享功能的C末端相似,可以诱发肺癌肿瘤发生和喀斯特监管元素提供其肺上皮细胞的特异性42,43。在不同的环境中,一个喀斯特水为等位基因是与WT相结合喀斯特,极品或喀斯特KI等位基因。自发性肺肿瘤的数量在喀斯特高水/吻,但同样低喀斯特水/ WT和极品KI;喀斯特水为老鼠。这种肿瘤抑制活动只能由一个完整的WT轨迹能够编码一个完整的喀斯特4同种型,因此,被一个类似的功能取代极品KI等位基因42。因此,现在有令人信服的证据表明,一个正常的WT Kras基因抑制和优先突变喀斯特4等位基因积极诱导肺肿瘤发生,分别在一个严格的组织特定的方式42- - - - - -44。事后看来,这也解释了早些时候vinyl-carbamate老鼠与一个治疗的观察喀斯特KO等位基因产生更多的更先进的比控制WT腺癌喀斯特老鼠45。
我们知道体细胞的发生喀斯特突变在肺癌中普遍存在46,从而使喀斯特的精确作用通路效应器的理解非常重要(图2所示⇑)。
事实上,突变在候选人效应器BRAF和PI3K的Ras激活下游通路在人类非小细胞肺癌已报告47,48。激活的突变BRAF最有可能刺激影响增殖蛋白激酶(MAPK)的途径。这个假设在肺具体测试,Tet-inducible,表达BRAFV600E突变体转基因46。在这只老鼠模型中,CCSP-rtTA;TetO7-BRAFV600E显示与bronchioalveolar肺腺癌癌的发展特性。细胞外signal-regulated激酶(ERK) 1/2 MAPK通路显著调节。在强力霉素,BRAF-mutant肺肿瘤消失在ERK1/2结合磷酸化的显著减少。此外,在活的有机体内使用特定的MAPK / ERK激酶(MEK)抑制剂诱导肺肿瘤回归。有趣的是,后者效果可以复制肺肿瘤CCSP-rtTA;TetO7喀斯特G12D老鼠46。这些结果表明,激活BRAF和喀斯特信号转换在同一MAPK通路,使这个途径肺肿瘤干预的潜在目标。这些结果证实了另一个独立研究中,液氧BRAFV600E等位基因在肺上皮细胞被激活通过鼻Adeno-Cre交付49,导致一个良性腺瘤的形成。然而,大多数这些后者病变没有进一步发展为腺癌和丰富nonprogressed腺瘤也表现出致癌基因诱导衰老的特征。结合BRAFV600E激活的损失Trp53或Ink4a /论坛领导,正如所料,标志着肿瘤进展为腺癌49。仔细调查期间喀斯特G12D驱动的肺肿瘤发生显示增加磷酸化的MAPK在MAPK拮抗剂Sprouty-2 (Spry-2)调节。当Cre / lox依赖Spry-2/ F;LSL喀斯特G12D老鼠用于肺肿瘤诱导,明显提高肿瘤负荷和发展中观察到喀斯特G12D;Spry-2- / -肿瘤。这清楚地表明肿瘤抑制基因活动期间Sprouty-2喀斯特依赖肺肿瘤发生损伤的Ras / MAPK信号级联50。
另一种方法来理解Ras下游效应的作用途径导致同时Cre /液态氧相关的删除Rac1/ F在激活喀斯特51。结果显示显著延迟喀斯特腺瘤形成的依赖Rac1- / -与Rac1+ / -病变,这表明喀斯特信号行为,至少在某种程度上,通过Rac1在肺肿瘤的发展。
另一个下游效应器(图2所示⇑),与RAS信号通路相互关联,phosphoinositide-3-kinase (PI3K)和下游目标,比如Akt蛋白激酶。在人类非小细胞肺癌体细胞突变激活PI3K已确定p110α催化亚基(编码PIK3CA)52。此外,选择性PIK3CA放大发现肺鳞状细胞癌53。因此,老鼠p110αTet-inducible表达式,生成的激活突变激酶结构域(H1047R),然后越过CC10 promoter-reverse四环素(rtTA)生成反式激活因子蛋白质CCSP-rtTA; TetO7PIK3CA (H1047R)老鼠。依赖在强力霉素诱导PIK3CA (H1047R) 14周,双转基因小鼠发达腺癌,随后强力霉素戒断3周后完全消失54。相反,完全消融PI3K活动通过删除其p85管理单元(编码Pik3r1和Pik3r2)导致了肺部肿瘤的数量急剧减少LSL喀斯特G12D;Pik3r2- / -;Pik3r1- / -老鼠54。许多在体外研究表明,Ras蛋白直接与p110α交互单元PI3K和引入特定的突变PIK3CA块这种交互55。为了研究Ras-p110α交互在活的有机体内及其对肿瘤发生的影响,Pik3ca- / -老鼠和交叉生成的喀斯特水为等位基因。这些Pik3ca- / -;喀斯特水为老鼠高度耐药对肺肿瘤形成,这表明Ras-p110α交互需要Ras-driven肿瘤发生55。
所有这些结果强调的重要性PI3K信号不仅对肺肿瘤诱导也维护。
相声Ras-MAPK之间转化生长因子(TGF) -β1及其II型受体(TGF-βRII)确实存在56,而TGF-β1涉及作为一个肿瘤抑制基因在人类肺癌57。为了测试这个假说在活的有机体内、肺肿瘤发生之后喀斯特水为;Tgf-β1+ / -与喀斯特水为老鼠。的喀斯特水为;Tgf-β1+ / -肺肿瘤出现更快的速度和相对较高的腺癌相比喀斯特水为老鼠。令人惊讶的是,腺癌,保留WT TGF-β1水平已经显著提高血管生成活动Tgf-β1+ / -腺癌。因此,TGF-β1是a善意的肿瘤抑制基因在肺肿瘤发生的发病和进展,但是还需要在同一时间肺tumour-induced血管生成和转移潜能58。
另一个关键球员驾驶人类非小细胞肺癌表皮生长因子受体信号、独立和上游的喀斯特(图2所示⇑)。事实上,突变表皮生长因子受体从这些中出现相互排斥喀斯特基因在人类非小细胞肺癌48。
大多数的突变表皮生长因子受体发生在该地区编码ATP结合口袋的受体酪氨酸激酶域在第18 - 21外显子59。更准确地说,这些突变发生在坐标系中删除外显子19日,消除一个守恒LREA(赖氨酸、精氨酸、谷氨酸和丙氨酸蛋白序列)主题(表皮生长因子受体▽),21外显子的L858R替换48。当这些老鼠脑中的特定突变工程利用Tet-inducible系统,两种类型的bitransgenic老鼠生成:CCSP-rtTA; Tet-O7-hEGFRL858R和CCSP-rtTA; Tet-O7-hEGFR▽。bitransgenic小鼠均表达了人类表皮生长因子受体突变肺II型肺泡细胞,经过几周的强力霉素政府开发支气管肺泡癌60,61年。然而,肿瘤的发展继续强力霉素应用时期延长超过4周,导致入侵腺癌的形成。然而,当强力霉素被撤回的缺失突变hEGFR导致肺癌的一个完整的回归60。总的来说,与人类腺癌肿瘤表型表现出显著的相似之处。这使得这些bitransgenic模型很好的研究工具EGFR-dependent NSCLC确认hEGFR▽和hEGFRL858R是显性的致癌基因能够诱导完全成熟的肺癌。另一个不同的表皮生长因子受体变异,变异III(八)在坐标系删除外显子2 - 7日,被发现在分析人类的鳞状细胞癌的5%,但腺癌62年。BitransgenicCCSP-rtTA; TetO7表皮生长因子受体八世老鼠腺癌16周后强力霉素的应用程序开发。肺肿瘤取决于EGFRvIII表达强力霉素撤出导致完整的肿瘤回归62年。所有表皮生长因子受体突变小鼠模型显示在非小细胞肺癌表皮生长因子受体活动的重要性和高度渗透在生产先进的腺癌。
因为磷酸酶的表达和tensin同系物删除从10号染色体(PTEN)通常是在NSCLC监管,一些老鼠模型中生成的Pten在支气管上皮细胞灭活63年,64年。PTEN是一种肿瘤抑制基因,可以通过阻断PI3K依赖serine-threonine激酶Akt激活(无花果。1⇑)。自Pten- / -小鼠胚胎致命,有使用液氧Pten等位基因(Pten/ F),加上CCSP-Cre转基因,目标Pten删除为支气管克拉拉细胞。然而,这些Pten/ F;CCSP-Cre老鼠没有任何异常的肺开发或表型异常即使随访≥12个月64年。这发生了翻天覆地的变化Pten/ F;CCSP-Cre等位基因是结合LSLKrasG12D。肺肿瘤发生明显加速比是单身LSLKrasG12D老鼠。喀斯特突变体,Pten缺乏更高级的腺癌类型的肿瘤通常是有多血管64年,这表明Pten损失与合作喀斯特突变在非小细胞肺癌。与这些结果的另一项研究的结果Pten失活在bronchioalveolar上皮与目标SP-C-rtTA; TetO7cre63年。强力霉素应用时在子宫内在E10-16胚胎发生,大多数老鼠出生后死于缺氧。肺部表现出受损的肺泡上皮细胞分化与整体肺上皮细胞增生。少数幸存的小鼠自发性肺腺癌发展。产后强力霉素应用在P21-27导致轻微的细支气管和肺泡细胞增生和细胞大小增加但没有杀伤力。大多数这些动物发达相比,腺癌WT控制。之前添加聚氨酯诱导更高的腺癌。有趣的是,大多数Pten- / -腺癌(33%),有或没有聚氨酯,显示自发Kras突变。后者再观察表明喀斯特活动配合的重要性Pten在NSCLC开发过程中损失。
最近的研究显示,大部分的非小细胞肺癌细胞的种系突变和受损的表现LKB1肿瘤抑制基因,丝氨酸苏氨酸激酶也被称为STK1165年。突变LKB1在Peutz-Jeghers综合征(睡衣)患者的特点是肠息肉(错构瘤)和上皮癌的发生率增加66年。然而,体LKB1突变已确定在一个小的一部分这些零星的肿瘤,如恶性黑素瘤、ovaria、乳腺癌和胰腺癌67年。一个例外是由肺癌LKB1失活非小细胞肺癌是一种常见的事件65年,68年。在腺癌突变被发现的最高数字65年和更频繁的腺癌喀斯特突变69年。有趣的是,肺癌没有睡衣中零星的癌症患者发病率最高67年。这表明LKB1的损失函数可能不是主要的遗传事件本身使肺肿瘤起始,而是一个重要的促进病变确保肿瘤进展。研究的结果Lkb1损失的背景条件LSLKrasG12D依赖在活的有机体内小鼠模型70年表明LKB1失活与喀斯特激活合作,建议LKB1的作用积极喀斯特下游通路的抑制因子。此外,单Lkb/ F老鼠没有显示出肿瘤表型。只有Lkb1/ F/ LSLKrasG12D非小细胞肺癌的小鼠表现出广泛的规模:多数是腺癌,但意外状癌和大细胞癌(LCC)也发生。此外,61%的腺癌发展为鳞状细胞癌转移但没有被检测到和LCC。相比p53/ F/ LSLKrasG12D和(Ink4a / Arf)/ F/LSLKrasG12D,Lkb1/ F/ LSLKrasG12D小鼠肿瘤高渗透和重要的高转移的数量。此外,没有鳞状细胞癌或LCC被检测到p53/ F/ LSLKrasG12D和(Ink4a / Arf)/ F/ LSLKrasG12D老鼠。结果表明,LKB1损失许可鳞状分化并促进转移,但这些是独立事件。腺癌的Lkb1/ F/ LSLKrasG12D小鼠减少pAMPK(磷酸化,adenosyl monophosphate-activated蛋白激酶)和pACCA(磷酸化,乙酰辅酶a carboxylseα-subunit)水平和激活mTOR(机械的雷帕霉素靶蛋白)通路(图2所示⇑)。可能LKB1损失影响非小细胞肺癌亚型分化通过离散路径71年。人类非小细胞肺癌的大型面板显示LKB1突变腺癌(34%)、鳞状细胞癌(19%)和LCC (16%)70年。伴随的突变p53和LKB1建议在非小细胞肺癌不重叠的角色。此外,重建人类非小细胞肺癌细胞株LKB1的显示抗肿瘤作用独立于其p53或(INK4A / ARF)状态70年。最后,失去LKB1表达肺泡腺瘤的增生,前体病变腺癌,表明早期LKB1的角色在腺癌发展失活72年。
不仅可以直接(epi)基因突变的癌基因和肿瘤抑制基因影响肺肿瘤发生过程中各自表达,但是小分子核糖核酸(microrna)也可以执行类似的角色。这些microrna类的小非编码rna转录调节目标mRNA的表达记录。为了成为活跃的小核RNA),主要的microrna的成绩单由核糖核酸酶进行催化裂解DICER1。在人类肺癌,DICER1活动的增加和变体规则的microrna的集群已经被观察到。对于后者,频繁的监管let-7 microrna的家庭以及upregulation mir - 17 - 92的报道73年。这mir - 17 - 92编码一组七个microrna转录成一个主记录。迄今为止,功能分析Dicer1和let-7一直在后台执行的喀斯特诱导非小细胞肺癌模型。一个条件删除Dicer1的背景LSLKrasG12D;Dicer1/ F老鼠让肿瘤发展的显著增加74年。然而,由于3′UTR区域喀斯特成绩单已被证明是let-7的直接目标75年,特别是容易增加let-7表达式喀斯特G12D肺肿瘤。事实上,两adenoviral鼻内的应用程序76年和慢病毒77年let-7 microrna的造成了严重的下降喀斯特G12D;p53- / -肺肿瘤。这些结果显示潜在的microrna的监管为NSCLC的维护的重要性。
为SCLC模型
与非小细胞肺癌,神经内分泌癌几乎是从来没有发现在自发或化学诱导小鼠肺癌。原因之一可能是,在这些小鼠肺癌p53和Rb基因突变的组合几乎从未发现,相反人类sclc的大部分。为了避免这种情况,一个基于Cre / lox删除的条件为Rb和p53等位基因是由气管内的与Adeno-Cre滴注法78年。3个月后,各种神经内分泌增生病灶发展通过近端和远端支气管。另外3个月后,这些早期病变进展到大规模肺肿瘤与SCLC的典型的组织学特征。有趣的是,一些早期病变类型仍然即使在广泛的SCLC的存在。结果的重要性来确定早期神经内分泌病变确实是SCLC前兆,如果是这样,然后额外的(epi)基因事件需要进步。Immunohistological性格化的成熟的肿瘤透露,他们确实与人类SCLC分享神经内分泌功能。所有神经内分泌分化标记,如降钙素相关基因蛋白(CGRP怎样),特异性神经元烯醇酶、synaptophysin,神经细胞粘附分子和achaete-scute homolog-1(火山灰)并不表示相同的方式在人类小细胞肺癌。此外,小鼠SCLC容易扩散至对类似的器官与人类SCLC发现78年。所有主要SCLC,以及他们的转移,都Rb和p53等位基因灭活。肿瘤,保留一个WT Rb基因都总是没有任何神经内分泌腺癌特征。因此,Rb最有可能的状态决定了如果肿瘤发生混合SCLC和非小细胞肺癌患者表型,如病人有时也被观察到79年。在Rb没有能找到肺肿瘤/ F老鼠,这表明,Rb损失不足以启动肺肿瘤发生78年和p53的额外的损失是必要的。
Rb不仅需要额外的基因事件损失,这些互补病变的性质也决定了哪些类型的肺癌将开发。例如,RB失活和Kras突变被发现时几乎从未在人类肺癌相同。此外,RB人类非小细胞肺癌的总体变异率非常低80年。正如我们所见,Adeno-Cre依赖激活的喀斯特广泛的肺上皮细胞导致完全非小细胞肺癌的发病。然而,类似的应用Adeno-Cre灭活RB(和p53)给神经内分泌SCLC完全不同。这些结果暗示,Rb损失在喀斯特没有函数驱动的肺肿瘤发生吗?当然不!当LSLKrasG12D转基因是结合Rb/ F和Rb的家人p130/ F等位基因为Adeno-Cre依赖肺肿瘤诱导,结果清晰地表明更进步的腺癌喀斯特G12DRb;- / -;p130- / -基因型相比,单喀斯特G12D81年。Rb损失和p130,尽管在较小程度上,导致喀斯特NSCLC的依赖。显然,在这种基因背景下喀斯特G12D否定任何Rb损失对神经内分泌分化的影响。另一个有趣的观察CC10-rtTA; tetO7cre; Rb/ F老鼠接受了强力霉素应用在早期胚胎发生,造成一个完整的Rb消融在所有支气管克拉拉细胞。然而,只有增加恶性细胞神经内分泌病变中发现这些老鼠,不影响克拉拉细胞内稳态可以观察到。与此相反,当所有三个Rb家族蛋白质(Rb, p107和p130)被截断SV40大t抗原灭活肿瘤蛋白(T121)CC10-T121老鼠,严重支气管增生与完整的所有克拉拉细胞发生去分化。这些结果表明,Rb可能特别需要确定神经内分泌细胞的命运,但是只有在一个严格的细胞和遗传背景。这一切从小鼠模型越来越多的证据表明非小细胞肺癌和小细胞肺癌做开发是不可能从类似的靶细胞。更合理的分开,异卵靶细胞可以发育成不同的肺癌,虽然每个主要还取决于具体的基因通路。
除了体细胞Rb/ F;p53/ FSCLC模型,另外两个肺癌模型也与肺神经内分泌肿瘤有关。一个模型利用bitransgenicCC10-hASH1; CC10-SV40大T进步的神经内分泌发育不良和积极与局部神经内分泌分化肺腺癌发展(通过表达pro-neural火山灰转录因子)和CC10表达82年。这些与神经内分泌分化腺癌相似人类非小细胞肺癌83年。其他模型用灭活的细胞周期蛋白依赖性激酶抑制剂p18 (Ink4c),即,一个肿瘤抑制基因在人类多发性内分泌瘤删除。p18自发性肺肿瘤的形成- / -,即+ / -老鼠最初导致腺瘤形成稳步增长,经过长时间的潜伏期> 1年,神经内分泌癌和腺癌的混合物84年。然而,这些混合肺肿瘤不同于那些在喀斯特开发的G12D通过他们的神经内分泌分化。最后,相比之下,Rb/ F;p53/ F这些小鼠肺肿瘤缺乏SCLC的典型的组织学和转移模式。
模型为鳞状细胞癌
人类肺鳞状细胞癌与吸烟密切相关,显示了不同的序列3变化在气道上皮细胞增生,化生、发育不良和癌原位对完整的浸润性鳞状细胞癌85年。然而,正常的人类肺或鼠标不包含鳞状上皮。只有在病理条件下鳞状分化(伴随着高角蛋白的表达水平,如角蛋白14 (K14))进行气道上皮的发生86年。这就解释了为什么自发SCC几乎从来没有发生在老鼠和只有少数小鼠模型报道鳞状细胞癌的发病主要是致癌物质后的应用程序。后者模型使用插管或气管内的甲基氨基甲酸酯87年或广泛的局部应用N-nitroso-methyl-bis-chloroethylurea和N-nitroso-tris-chloroethylurea88年,89年。整个光谱的不正常鳞状上皮表型观察,但是只有在受到纯系小鼠(NIH瑞士、A / J, Balb / cJ, FVB / J和SWR / J),而不是在其他(C57BL / 6 J, AKR / J - 129 / svJ)89年。除此之外,特定位点SCC敏感性可以通过链接分析发现在几个老鼠菌株89年。另一项研究报道完全不同的方法试图通过组成型表达的人类K14诱发鳞状细胞癌通过CC10-hK14老鼠90年。虽然CC10-hK14老鼠FVB / J背景和hK14在支气管上皮容易表达,只有前体病变不同可以观察到从增生、鳞状上皮化生90年。显然,增加K14表达和出现鳞状分化本身并不足以产生全面先进的鳞状细胞癌。然而,正如我们已经描述了,到目前为止只有LSLKrasG12D;Lkb1/ F体为非小细胞肺癌小鼠模型已经能够产生先进的鳞状细胞癌。LKB1失活导致侵略性和转移性肿瘤的60%的鳞状或混合鳞状组织学70年,尽管前体病变进行航空也不见了。
平移肺癌小鼠模型的治疗研究
异种移植模型被广泛用于肺癌治疗的临床前测试。这主要表现在人类肺癌细胞系后免疫缺陷小鼠皮下注入。更多的首选方法,然而,将orthotopical移植的人类肺肺肿瘤细胞在自然环境。迄今为止的结果表明,异种移植模型有不良记录的准确预测抗肿瘤药物的临床疗效。因此,一个合理的问题是是否自发对肺癌和转基因小鼠模型将更有用的作为临床前药物测试的工具。很明显,老鼠和人类之间有肺生理上的差异,但一些描述的小鼠模型,我们已经有一个显著的表型相似之处,结合基因签名非常类似于人类非小细胞肺癌35和SCLC91年。重要的是,转基因老鼠model-derived肿瘤发展一个先天免疫的环境,因此,使用所有tumour-stromal交互,如血管生成和退化的组织矩阵。
事实上,第一个报告已经出版,显示非常有前途的预测能力的小鼠肺肿瘤治疗研究的模型,我们将讨论两种。
不过,首先,我们要简单地址从研究的一些发现/ J小鼠易感carcinogen-induced肺肿瘤发生。使用标准化carcinogen-induction协议/ J小鼠产生一个高度可再生的定义良好的肺腺癌92年。因此,一个/ J小鼠被广泛用来测试化学干预的疗效。例如,50 - 60%的肿瘤质量减少腺癌治疗后获得cis-platinumin结合胃复安和/或吲哚美辛,而腺瘤增长不受影响93年。由于应变/ J肺肿瘤模型已经证明了他们的效用测试化学疗法,他们也被广泛用于评估化学预防协议94年。广泛的chemopreventive代理,如通过转移酶抑制剂,isothyocynates,非甾体类抗炎药、糖皮质激素以及绿茶和胡萝卜素等天然产品只是到目前为止已经测试的一些示例95年。在最好的情况下,肺肿瘤生长被50 - 60%受损94年。引入p53和p16ink4a/ p19东盟地区论坛突变等位基因为应变/ J小鼠造成的,正如所料,提高肺部肿瘤外显率而不是一个戏剧性的差异在化学预防的敏感度96年。为了揭开无疑的复杂的遗传性状,导致chemopreventive效应/ J菌株,第一个基因签名等chemopreventive代理绿茶和budenoside使用基因表达阵列97年,98年。
我们已经描述了两个模型NSCLC喀斯特的连续致癌活性29日或表皮生长因子受体61年先决条件的肿瘤维护。这不仅表明,肿瘤生长严重致癌途径取决于各自的活跃,但它也强调这些致癌途径作为治疗目标的有效性。肿瘤直接干预研究酪氨酸激酶抑制剂(TKIs)对表皮生长因子受体突变被证明是非常有效的在几个hEGFR转基因小鼠模型。TKIs如吉非替尼、埃罗替尼及hki - 272领导完成肿瘤回归60- - - - - -62年。此外,长期治疗与人性化anti-hEGFR抗体(西妥昔单抗)引起了强烈的肿瘤回归60。将需要进一步研究探讨通路确定敏感性和抗表皮生长因子受体相关TKI干预措施。其他非小细胞肺癌小鼠模型也被用于靶向治疗。首先,诱导表达的PI3K p110α催化亚基(PIK3CA)突变激酶结构域(H1047R)CCSP-rtTA; TetO7PIK3CA (H1047R)老鼠,诱发腺癌。治疗的肺肿瘤NVP-BEZ235,双重pan-PI3K和哺乳动物雷帕霉素靶(mTOR)抑制剂,导致明显的肺肿瘤回归。有趣的是,当这一个代理NVP6-BEZ235测试在肺肿瘤CCSP-rtTA; TetO7喀斯特G12D老鼠,没有观察到的回归。然而,在NVP-BEZ235 - 142886,结合MEK抑制剂进行重要的回归喀斯特G12D肿瘤发生54。显然,两个主要的RAS(鼠肉瘤病毒癌基因蛋白同系物)下游效应器通路(图2所示⇑)需要受损为了得到一个不可逆的回归在Ras突变的非小细胞肺癌。
直接针对RAS迄今为止广泛成功的肺癌治疗。许多小分子对Ras功能已经测试并通过转移酶抑制剂是最明显的例子这些失败的尝试99年,One hundred.。一个解释可能被发现,只有KRAS4B farnesylated KRAS4A。但不是其对碘氧基苯甲醚最近的结果与肺癌小鼠模型,正如我们前面描述的那样,强烈建议实际上KRAS4A而不是KRAS4B驾驶非小细胞肺癌的发病。虽然我们还得看看KRAS4A确实是重要的人类非小细胞肺癌,一个人可以想象的重要性喀斯特KRAS4A功能抑制小鼠模型的测试42。
使用优化转基因小鼠模型为肺癌治疗研究需要复杂的非侵入性工具遵循肿瘤发展和对治疗的反应。测量肿瘤大小作为时间的函数是最明显的的方法和现有的技术,如正电子发射tomography-computed断层扫描或磁共振成像用于使用小动物54,61年。然而,这些技术是耗费时间,使他们不太适合高吞吐量。其他敏感和可再生的技术现在被广泛用于测量基因表达和肿瘤生长在活的有机体内:荧光成像和生物发光101年,102年。荧光蛋白的表达可以耦合到致癌基因转基因小鼠模型中促进肿瘤增长的荧光成像。生物荧光,荧光素酶的转基因表达允许精确的纵向监测和肿瘤的好量化质量已被证明的LSL喀斯特肺肿瘤模型103年。可行的成像技术将极大地增强小鼠模型的准确性和重现性。
我们展示了一些治疗小鼠模型的研究的应用前景。然而,人们不应该忘记,鼠标转基因模型是非常简单的近似人类的疾病,尽管一些体细胞模型的人类肺癌有惊人的相似之处。肺肿瘤在小鼠模型通常是由有限,他们仍然依赖路径定义。是否人类肺癌显示类似的依赖持续的活动启动致癌基因还有待观察。遗传工程小鼠模型的力量,它可以让我们了解这种特定基因损伤的重要性在治疗肿瘤生长。因此,它可以是一个有价值的治疗更有效的临床前研究的第一步。
肺癌干细胞的起源
初始突变细胞接受一系列的各种基因改变之前,他们最终获得完整的肿瘤发生的能力104年。不同的肿瘤发展的早期阶段已确定为人类鳞状细胞癌和腺癌6建议改变了支气管和肺泡细胞本身可以作为肺癌细胞的起源。我们已经表明,这已被证实对大多数人来说,但不是全部,为肺癌的转基因小鼠模型。非小细胞肺癌的结果以及SCLC模型70年,78年,81年表明,这两种不同类型的基因突变和程控这些病变控制初始事件发生肺癌的发展。然而,不确定性的存在对肺癌细胞起源的本质。了解这些细胞可以产生重要的影响,不仅对我们了解肺癌生物学还通过提供我们肺癌治疗的新目标。多年来它已经知道人类腺癌和肺癌SCLC包含一小群细胞(< 1%),能够在软琼脂形式殖民地105年。移植到老鼠无胸腺的裸体后,选择软琼脂殖民地被证明产生原始的原发性肺癌肿瘤具有类似的特性105年。因此,人类肺癌显然由异构的细胞类型的只有一小部分能完全恢复肿瘤。人们很容易将肺癌细胞的再生或单独使用子集“肿瘤干细胞”(CSC)。作为一个CSC意味着他们分享功能或直接来源于突变,改变了正常的干细胞。正常干细胞的特性是什么使他们这样理想的肿瘤发生的目标?干细胞被定义为罕见,长寿细胞的主要属性包括自我更新能力,广泛的扩散通过的细胞,并通过代多功能能力成熟,终末分化细胞106年,107年。自我更新允许维护一个未分化的干细胞池在宿主的生命周期和其长寿使二者一起积累足够的遗传病变的理想候选人需要启动肿瘤发生。正常干细胞的多药耐药性是一个额外的属性,保证他们的长寿面对有毒代理人,包括许多著名的药物用于治疗肺癌。功能多药耐药性通常是由过度表达的腺苷triphosphate-binding磁带(ABC)转运体释出药物108年。事实上,在描述了ABC转运蛋白的表达在单独使用子集,治疗肿瘤的药物抗性的可能的原因108年。后者的观察也可以特别感兴趣的人类肺癌化疗和放疗后复发以来在人类肺癌是常见的109年。定义chemoresistant CSC人口可能是一个潜在来源的肿瘤复发,因此本身成为一种有价值的治疗目标。
然而,直到最近,相似性癌细胞和正常干细胞的想法导致上皮肿瘤内存在一个小的人口CSC或“肿瘤起始细胞”108年。二者被定义为一种罕见的未分化细胞致瘤的肿瘤起始,负责维护和传播106年,108年。这些细胞在许多类型的癌症已确定分类细胞亚种群基于表面标记表达模式,并通过移植研究动物模型使用前瞻性孤立的肿瘤细胞。他们的存在是第一次有记录的白血病110年,111年。这之后,他们已确定在乳腺癌112年和胶质母细胞瘤113年。最近,“肿瘤启动细胞”已确定在多种肿瘤包括前列腺癌、结肠癌、胰腺癌和肝癌、黑色素瘤和其他几个肿瘤类型114年。这些研究表明,这些细胞提供了一个无限更新潜力,他们可以接受分化形成肿瘤的免疫缺陷小鼠概括原始肿瘤的异质性效率高115年。然而,目前尚不清楚是否存在二者在一个坚实的肿瘤。然而,癌症的个体发生的另一个可能性是所谓的随机模型。这个模型预测癌症的发病和进展从多个转换祖细胞导致多克隆肿瘤,在每个subclone可以,原则上,重建一个完整的肿瘤116年。因此,后者的模型预测,每一个转换,未分化的细胞有相同的致瘤的能力和选择的肿瘤将包括最主要的积累117年。一个相对高的比例从单个肿瘤细胞,因此,可以发展成成熟的肿瘤异种移植后免疫缺陷小鼠118年。
因此,有证据表明不同的肺癌细胞群,这需要更好的理解正常的支气管和肺泡组织是如何组织为了确定肺干细胞。
肺组织在很大程度上是内生的形成和更新,现在人们普遍认为,循环间质肺上皮细胞的祖细胞的形成是罕见的,而不是生理的相关性119年。肺干细胞研究进展的缓慢,而在其他器官相比,由于解剖和功能与细胞的异质性和复杂性低流动率的上皮细胞。只有在受到环境和营养因素的肺上皮细胞显示其流动率大幅增加。这是在矛盾不断迅速恢复肠道的组织和造血的起源。这些快速更新组织有一个古典分层组织的干细胞,分化的祖细胞和他们的后代。肺系统,然而,包含各种各样的模有组织的细胞,(兼)祖细胞和终端分化的细胞结构上进行气道上皮细胞分离和气体交换肺泡地区120年。这些不同的肺上皮细胞主要是通过呼吸道损伤识别实验。各种有毒的药物被用来触发特定的肺上皮细胞的选择性损伤反应之后,他们增加增殖率在活的有机体内通过(3H]胸苷或bromodesoxyuridine (BrdU)。在脉冲/追逐实验,只有干细胞自我更新的保留这个标签在较长时间内由于缓慢的营业额和所谓的标签保留细胞(荣誉奖)。
虽然远端肺上皮由克拉拉、纤毛和神经内分泌细胞,只3H]标签Clara-like细胞能够增殖并产生被发现新创差异化的克拉拉细胞和纤毛细胞与臭氧气体接触后最初纤毛细胞受损121年。这提供了证据,克拉拉细胞是兼性祖细胞,但它没有提供证据Clara-cell人口中干细胞的层次结构。另一个毒剂,萘,诱发消融的几乎所有nonciliated细支气管上皮克拉拉细胞由于选择性毒素的代谢活化。这治疗的结果是两个不同的种群的扩散领头地区局部神经内分泌的身体(nebs3级)远端气道,即Clara-like细胞CC10表达和肺神经内分泌细胞(PNECs) CGRP怎样表达122年。两naphthalene-resistant Clara-like细胞(名为变体或vClara细胞)和PNECs能够维持上皮更新及其本地化nebs3级被认为代表一个干细胞利基123年- - - - - -125年。vClara细胞耐药萘受伤因为他们不表达酶Cyp2f2,这是细胞色素P450家族的一员,将萘转换成一种有毒的化合物。除此之外,vClara细胞还能流出药物通过耐多药受体和呈现一个人口表型126年。
转基因小鼠模型的单纯疱疹病毒胸苷激酶(TK)基因表达的控制下CC10启动子(CC10-TK)是用来测试是否PNECs vClara人群表示与多能干细胞池气道上皮修复的能力127年。在这个模型中CC10-expressing (如。vClara和正常的克拉拉)细胞可以有选择地切除小鼠的航空公司中通过急性更昔洛韦。研究再生上皮显示CC10-expressing细胞的消融导致PNEC扩散和增生,但他们无法有效增殖分化纤毛上皮127年。PNEC增生独立于NEB-associated vClara细胞可能代表一种代偿机制的设置,气道总干细胞消融128年。因此,这些研究导致的结论是,一个族群CC10-expressing细胞,但不是PNECs,可能代表干细胞修复所需的克拉拉细胞损伤小鼠气道远端或这些细胞可能需要支持干细胞产生新的克拉拉细胞127年。
第二个上皮干细胞利基类似萘治疗后已经被确认123年,129年。领头的一个族群附近发现了克拉拉细胞终端支气管,在bronchioalveolar管结(BADJs)航空公司终止,形成肺泡,只有极少数人nebs3级123年,124年。这些细胞被称为bronchioalveolar干细胞(过时)129年。他们co-express CC10 SP-C,这是一个主要的分泌化合物II型肺泡细胞,但在低水平表达的早期胚胎肺上皮细胞130年。然而,双重表达CC10 / SP-C细胞胚肺底没有被发现,所以很可能这些过时在快速扩张发展出生后肺上皮组织。
这双正(CC10和SP-C)人口是静止在正常肺131年。过时发现表达本来就和CD34细胞表面标志,这是建立在其他老鼠组织干细胞表面标记132年。可行(本来+ CD34 +)过时被流式细胞术能够独立排除血液(CD45 +)及内皮细胞(CD31 +)129年。他们似乎是至关重要的再生终端细支气管和肺泡上皮组成部分。此外,过时几个段落的能力在体外而维护他们(CC10 + / SP-C +)状态或发展成克拉拉(CC10 + / SP-C -),肺泡II型(CC10 - / SP-C +)或肺泡I型(SP-C + / aquaporin5 +)细胞。因此,过时被证明是多能干细胞分化和自我更新的能力。目前,我们仍然不这些细胞功能是否一样真正的干细胞的自我更新和multipotency证据在活的有机体内然而,初步的。显然更好的描述过时需要识别更多独特的表面标记,已经由几个孤立的研究中显示133年,134年类似或相同但也探讨如何vClara细胞和过时。事实上,到目前为止所有的证据表明vClara和过时的候选支气管干细胞和正常的克拉拉细胞是兼性的细胞。
另一个假定的肺干细胞人口被发现通过使用血清培养系统主要新生儿肺细胞。CC10殖民地产生积极的,本来但是为SP-C和CD34阴性。此外,细胞被发现表达octamer-binding转录因子4 (Oct4)和stage-specific胚胎antigen-1。BrdU脉冲追踪实验表明slow-cycling标签保留Oct4阳性细胞在BADJ本地化135年。这些Oct4阳性细胞能够分化成肺泡类型2或1细胞。再次还有待观察相似这个候选人支气管干细胞和其他两个候选人。研究发现CSC在人类肺癌大多是直接从初级肿瘤隔离不同的细胞群。到目前为止,没有明确的健壮的标记特征,可以只与单独使用CSC数量。这些结果在别处了136年。此外,由于没有已知的人类本来就相同器官,它变得明显,过时而且vClara细胞需要进一步描述为了找到它们的人类。最近,它也被证明人类小细胞肺癌和非小细胞肺癌含有小数量的未分化细胞表达细胞表面标记prominin-1 (CD133)137年。发现Cd133表达cancer-primitive细胞以及神经正常细胞,内皮细胞、上皮细胞和造血的血统138年。肺癌CD133阳性细胞作为肿瘤领域能够无限期生长在特定的培养条件和他们可以生成肿瘤异种移植免疫功能不全的老鼠,原始肿瘤表型相同,而CD133 -细胞没有137年。肺癌包含一个罕见的人口CD133阳性癌症干细胞样细胞能够自我更新并生成无限nontumorigenic细胞的后代137年。观察药物抗性在肿瘤领域,并与ABC转运蛋白的表达增加有关胚胎干细胞标记Oct4和NANOG。有趣的是,在正常小鼠萘治疗之后,一种罕见的CD133阳性细胞的数量增加。因此,它会很容易认为,那些扩大CD133阳性细胞是过时的。需要后续研究来证实这和一些其他发现鼠标模型转化为一个更好的人类肺CSC的描述。
支气管干细胞群可能肺癌祖细胞的理想人选。随着这些肺干细胞已经具有自我更新的能力,他们是优秀的候选人有害突变的积累。事实上,越来越多的证据已经过时,这确实是如此。
细胞表达SP-C和CC10首次描述的前驱病变小鼠肺肿瘤由致癌喀斯特33。表达这些双阳性细胞的致癌蛋白喀斯特诱发他们的扩散在活的有机体内33。此外,在naphthalene-treated诱导肿瘤发生LSL-KrasG12D诱发小鼠肿瘤的大小和数量的增加129年。此外,喀斯特G12D表达导致过时在体外扩张并生成BADJ增生在活的有机体内129年。同样,从p38α过时MAPK- / -老鼠也高度敏感喀斯特G12D全身的肺肿瘤发生,导致一个不成熟和hyperproliferative肺上皮细胞,从而允许早期感应和更快的发展为腺癌139年。废除主要信号通路被证明对过时的扩散和维护非常重要。条件删除PTEN或PI3K在老鼠身上增加数量的过时和倾向肺肿瘤的发展63年,140年。在p27- / -老鼠,过时扩大第一BADJ和进步通过发育异常增生,腺癌和腺瘤141年。相比之下,删除Bmi1抑制肿瘤发生通过抑制过时的扩张142年。综上所述,这些研究表明,过时可能发挥作用在肿瘤发生的起始,但仍然没有直接证据的初始目标细胞本身。事实上CSC可能起源于支气管干细胞,但我们不能排除他们发展的细胞,如PNECS和克拉拉细胞各有了自我更新的能力作为一个致癌突变的结果。除了收购遗传和表观遗传突变,肿瘤细胞及其微环境之间的相互作用(也称为利基市场,由基质、炎性细胞和招募了脉管系统),对肿瘤发生的过程有深远的影响。越来越多的证据表明,肿瘤增长能否持续不仅通过罕见CSC的数量也占主导地位(子)克隆或两者的混合物117年。虽然描述CSC的肺癌的重要性是明显的重要性,它将过于乐观地说,针对CSC的治疗肺癌。最有可能需要结合针对不同人群的长期影响肿瘤生长,防止复发。
视角和新方向
小鼠模型的使用是帮助我们理解肺癌生物学发现关键的分子途径控制每个阶段的肿瘤形成和发展。这将是重要的调整为每个不同的人类肺癌小鼠模型类型。全基因组表达基因组杂交分析和进展将有助于跨物种的相似性分析癌症信号通路管理两个老鼠和人类肺部肿瘤发生。这些研究将为抑制剂的设计非常有用,它可以损害肺部肿瘤生长,可以在临床前小鼠模型进行测试。新的、更好的适应小鼠模型和完整的人类基因可能导致人性化的发展概括人类肺癌的小鼠在更高的学位32。飞速发展的肺癌基因组分析将揭示小说许多额外的突变,可以补充为肺癌小鼠模型。
转移小鼠模型对非小细胞肺癌和小细胞肺癌已经很完善,这将是一个挑战,使用这些模型来解决的机制肺癌化疗通常是耐火材料——和/或放射治疗。定义早期肺癌的发展结合非侵入性成像技术在这些小鼠模型将是非常有用的化学预防试验协议。条件肺肿瘤模型的另一个有趣的方面是,他们可以为肺癌收益率有前途的生物标志物。高级质量spectromy-based蛋白质组学将允许审查小鼠血清蛋白质组的分析,在正常和癌变的条件下。有一个明确的需要早期肺癌的临床相关的生物标志物和体细胞为非小细胞肺癌和小细胞肺癌小鼠模型均显示出明显的肺肿瘤发展阶段。结合基因表达分析和蛋白质组学的这些早期的小鼠肺肿瘤可能会导致新发现的,相关的生物标记物,可以转化为诊断使用。一个重大的挑战不会候选生物标志物的发现,但评估患者样本通过适当的验证。
小鼠遗传学的非凡力量将继续帮助我们确定肺部肿瘤选择性靶向前体细胞的肺上皮细胞隔间。候选人前体细胞可以标记与报告基因可以跟随他们是否像癌症干细胞在肿瘤生长。
很大的进展取得了在人类肺癌小鼠模型(表1所示⇓,图3⇓),但是仍然需要进一步描述对于大多数非小细胞肺癌和SCLC模型。同样重要的是要注意,小鼠模型的发展为鳞状细胞癌明显缺乏。更好的肺鳞状细胞癌的细胞起源的知识,一起确定最适合遗传病变的原因这种肺癌的类型,可能会促进适当的小鼠模型的设计。
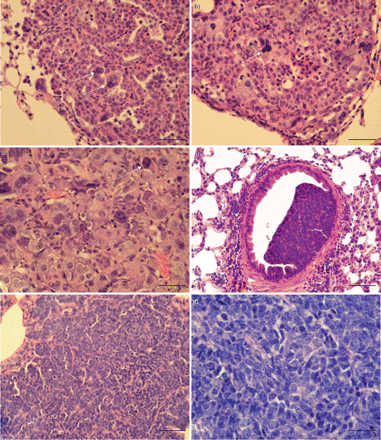
代表肺肿瘤小鼠模型的先进nonsmall细胞肺癌和小细胞肺癌。的进展腺癌Trp53/ F;LSLKrasG12V老鼠Adeno-Cre治疗后3个月所示(a - c;未公开的数据)。)腺癌中,一些肿瘤细胞不规则的原子核(箭头)。b, c)与pleiomorphic核进一步先进的显示肿瘤细胞腺癌(白色箭头)和巨细胞(黑色箭头)。d)早期神经内分泌增生性病变支气管内Rb/ F;Trp53/ F老鼠Adeno-Cre治疗后3个月,再三个月后,发展成小细胞肺癌(e, f)。注意邻近肿瘤细胞的核的特征造型和稀疏的细胞质。Haematoxin和伊红染色。a、b、d、e) = 25μm规模酒吧。c、f) = 10μm规模酒吧。
最后,我们已经看到,肺癌小鼠模型已经帮助我们进一步了解基本生物学和测试肺癌治疗肺癌。这些模型仍然有许多承诺应扩大在这些字段和通过寻找肺癌诊断的新指标。结果无疑将履行这些承诺,是对抗人类肺癌的重要性。
支持声明
s . de Seranno由博士后研究人员支持格兰特(杜Instiut国家癌症,巴黎,法国)和r . Meuwissen支持的另一幅作品《年轻Chercheur奖学金(INSERM,巴黎)。
感兴趣的语句
没有宣布。
脚注
本系列之前的文章:1号:德魏夫W, Stroobants年代,酵母J,et al。集成的PET / CT在nonsmall细胞肺癌的分期:技术方面,为肺癌切除术。欧元和J2009;33:201 - 212。2号:Rami-Porta R, Tsuboi m . Sublobar切除的肺癌。欧元和J2009;33:426 - 435。3号:威廉姆斯,Lam B, Sutedja t .近端早期肺癌诊断和治疗。欧元和J2009;33:656 - 665。4号:Sculier j], Moro-Sibilot d一线和二线治疗先进nonsmall细胞肺癌。欧元和J2009;33:916 - 930。5号:van Tilburg PMB,斯塔姆H, Hoogsteden HC,et al。肺癌患者的术前肺评价:文献之回顾。欧元和J2009;33:1206 - 1215。6号:Brambilla E, a Gazdar肺癌的发病信号通路:路线图疗法。欧元和J2009;33:1482 - 1494。7号:霍法我,Lazar Z, Gyulai N,et al。呼出肺癌的生物标志物。欧元和J2009;34:261 - 275。8号:托马斯•RK Ocak年代,Sos毫升,等。高通量分子分析肺癌:洞察生物学和潜在的临床应用。欧元和J2009;34:489 - 506。9:JK, Liloglou T, Niaz。et al。EUELC项目:多中心、多用途研究调查早期非小细胞肺癌,为正在进行的合作建立一个生物。欧元和J2009;34:1477 - 1486。10号:Demedts本土知识,维尔马伦肯塔基州,范Meerbeeck JP。小细胞肺癌治疗extensive-stage:现状和未来前景。欧元和J2010;35:202 - 215。
- 收到了2009年8月4日。
- 接受2009年8月10日。
- ©人期刊有限公司
引用
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}