





文摘
糖皮质激素是最有效的抗炎治疗哮喘。在哮喘炎症的特点是增加多个炎症基因的表达受炎性转录因子,如核能factor-κB激活蛋白1,结合并激活共激活剂分子乙醯化核心组蛋白和基因转录开关。糖皮质激素抑制多个在哮喘气道炎症基因被激活,主要是通过改变组蛋白乙酰化作用的激活炎症基因通过绑定的糖皮质激素受体辅活化因子和招聘组蛋白脱乙酰酶2的激活转录复杂。
激活糖皮质激素受体也绑定到特定基因的启动子识别网站为了激活转录,导致分泌的抗炎蛋白,增殖蛋白激酶phosphatase-1等抑制增殖作用蛋白激酶信号通路。糖皮质激素受体也可能与其他识别网站交互抑制转录,例如多个基因与它们的副作用。
在某些抗类固醇哮喘反应患者,糖皮质激素受体信号通路异常。在慢性阻塞性肺疾病患者和哮喘病人吸烟,组蛋白脱乙酰酶2是明显受损由于氧化/ nitrative压力,因此炎症是抗糖皮质激素的抗炎作用。
这些新发现的治疗意义进行了讨论。
系列”信号和转录调节炎症和免疫细胞:在肺重要性生物学和疾病”
编辑K.F.涌和贝聿铭爱德考克
数字4在本系列
糖皮质激素(也称为糖皮质激素、糖皮质激素或类固醇)是目前最有效的抗炎治疗哮喘和现在已经成为所有患者的一线治疗持续性哮喘和其他一些炎症和免疫疾病。理解的分子机制有重要进展,糖皮质激素抑制炎症有效在哮喘、基于最近的事态发展在理解基因转录的基本机制和细胞在炎症信号1,2。这种新理解这些分子机制也有助于解释糖皮质激素能够关闭多个炎症通路,但仍然是一个安全的治疗。它还提供了洞察为什么糖皮质激素抗类固醇哮喘反应患者无法工作,慢性阻塞性肺疾病(COPD)和囊性纤维化3。
糖皮质激素的主要作用是关闭多个炎症基因(编码细胞因子、趋化因子、粘附分子,炎症酶,受体和蛋白质)在炎症过程中被激活。他们有额外的影响抗炎蛋白的合成,同时后基因的影响。已经有相当大的兴趣在糖皮质激素如何影响信号的转导途径激活的炎症。
炎症的分子基础
慢性炎性疾病,如哮喘和慢性阻塞性肺病,涉及许多炎症和免疫细胞的渗透和激活,释放多种炎症介质相互作用和激活结构细胞的炎症。这些疾病炎症明显不同的模式,不同的细胞和介质的参与4,5,但是所有的特点是多个炎症基因的表达增加,其中一些是常见的炎性疾病,而另一些更具体的一个特定的疾病。这些炎症基因由促炎症控制转录因子,如核转录因子(NF) -κB和激活蛋白(美联社)1,这成为激活炎症过程。这些炎性转录因子激活炎性疾病和发挥重要作用在放大和延续炎症过程。因此,NF-κB被激活在哮喘患者的气道和慢性阻塞性肺病患者6,7。更多针对疾病的蛋白质更容易受到特异性转录因子,如核转录因子的激活t细胞,调节t淋巴球的某些细胞因子基因8或GATA-3,调节2型辅助细胞的分化和表达细胞因子在过敏性疾病9。
炎症基因表达的分子通路参与监管正在划定,越来越清晰,染色质重塑中起关键作用的转录控制基因。刺激炎症基因开关通过改变染色质的结构基因,而糖皮质激素逆转这一过程。
染色质重塑和基因表达
染色质的DNA和基本的蛋白质被称为组蛋白,提供结构的主干染色体。长期以来人们一直认为组蛋白发挥重要作用在调节基因的表达和确定哪些基因转录活跃,哪些抑制(沉默)10。染色质结构是高度组织由于近2米的DNA必须挤进每一个细胞核。染色质是由核小体,颗粒组成的146个碱基对的DNA伤口周围近两倍的八聚物两个分子的每个核心蛋白质组蛋白H2A、H2B, H3和H4。自1990年代中期以来,它已经可以阐明如何表达和基因的镇压与改造这种染色质结构的酶改性的核心组蛋白蛋白质,特别是通过乙酰化作用。每个核心组蛋白有着悠久的氨基端富含赖氨酸残基的尾巴,这可能成为乙酰化,从而改变组蛋白核心的电荷。在静息细胞,DNA是在核心组织蛋白,不含酶RNA聚合酶II的绑定,激活基因转录和mRNA的形成。这种构象的染色质结构被描述为关闭,与抑制基因表达有关。基因转录发生只有当染色质结构开放,解除的DNA, RNA聚合酶II和基底转录复合体可以绑定到DNA启动转录。
组蛋白乙酰转移酶和辅活化因子
当炎性转录因子,如NF-κB、被激活,它们绑定到特定识别DNA的序列,随后与大共激活剂分子,如cAMP-response-element-binding蛋白(分子)结合蛋白(CBP), p300和p300 / CBP-associated因素(pCAF)。这些共激活剂分子作为控制基因转录的分子开关,都有内在的组蛋白乙酰转移酶(HAT)活动11,12。这导致核心组蛋白的乙酰化,从而减少他们的指控,它允许休息的染色质结构将关闭构象激活开放形式12。这将导致解除的DNA,和绑定TATA-box-binding蛋白质和RNA聚合酶II和相关因素,然后启动基因转录。这所有基因的分子机制是常见的,包括那些参与分化、增殖和活化的细胞。当然这个过程是可逆的,和乙酰化组蛋白脱乙酰作用与基因沉默相关。这是由组蛋白去乙酰酶抑制剂(hdac),充当辅阻遏物和其他辅阻遏物蛋白质随后招募。
这些基本机制已经被应用于理解炎症的调控基因疾病,如哮喘和慢性阻塞性肺病13。在人类上皮细胞,激活NF-κB,通过公开细胞炎症信号如白介素(IL) 1β,肿瘤坏死因子(TNF) -α或内毒素,结果在特定的赖氨酸残基的乙酰化组蛋白H4(另一组蛋白似乎不那么明显或迅速乙酰化),这与基因编码的炎性蛋白的表达增加,如集落刺激因子(gm - csf)14。
糖皮质激素受体
糖皮质激素容易扩散通过细胞膜,与糖皮质激素受体结合(GRs)在细胞质中。细胞质GRs通常绑定到蛋白质,称为分子伴侣’,如热休克蛋白90和FK-binding蛋白质,保护受体和防止核本地化覆盖受体上的网站需要传输整个核膜进入细胞核20.。人类GR单个基因编码,但一些变体现在承认,由于替代翻译起始转录剪接和替代21。GRα结合糖皮质激素,而GRβ或者拼接DNA结合的形式,但不能激活糖皮质激素。GRβ展品GRα相比非常低水平的表达22。一直在与类固醇抗GRβ对碘氧基苯甲醚哮喘23,尽管GRβ能否有功能意义受到质疑的很低水平的表达GRα相比24。
GRs也可能被磷酸化和其他修改,这可能改变糖皮质激素的反应影响配体结合,核易位,反式活肤功效,蛋白质-蛋白质之间的关系或代数余子式的招聘25,26。例如,有许多的丝氨酸/苏氨酸残基的n端结构域GR可能被各种激酶磷酸化。
一旦糖皮质激素有一定会GRs,受体结构的变化导致分裂的分子伴侣蛋白,从而使核本地化GR信号。这导致激活GR-corticosteroid复杂的快速运输到细胞核,它绑定到特定序列的DNA corticosteroid-responsive基因的启动子区域称为糖皮质激素反应元素(格蕾丝)。两个GR分子聚集在一起,为GRE和绑定,导致基因转录的变化。经典交互与格蕾丝GRs导致基因转录增加(反式激活),但消极的GRE网站也被描述,在绑定GR导致基因的抑制(独联体镇压)(图。1⇓)27。很少有证据确凿的例子-格蕾丝,但一些相关的皮质类固醇的副作用,包括基因调节hypothalamopituitary轴(proopiomelanocortin和-因子),骨代谢(骨钙素)和皮肤结构(角蛋白)。
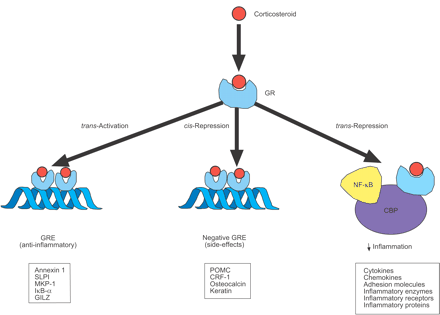
糖皮质激素可能通过多种方式调节基因的表达。糖皮质激素进入细胞糖皮质激素受体结合(GRs)在细胞质中,把原子核。GR为糖皮质激素反应元素绑定(格蕾丝)steroid-sensitive基因的启动子区域,这可能编码抗炎蛋白。一般较少,GR为相互作用-格蕾丝抑制基因,尤其是那些与糖皮质激素的副作用。核GRs也与共激活剂分子,如cAMP-response-element-binding-protein-binding蛋白质(CBP),由促炎症激活转录因子,如核转录因子(NF) -κB,因此关掉炎性基因激活这些转录因子。SLPI:分泌leukoprotease抑制剂;MKP:增殖蛋白激酶磷酸酶;NF-κB IκB-α:抑制剂;GILZ:激素性亮氨酸拉链蛋白质;POMC: pro-opiomelanocortin; CRF: corticotrophin releasing factor. ↓: decrease.
类固醇诱导性基因转录
糖皮质激素产生影响响应细胞通过激活GRs为了直接或间接地调节目标基因的转录。每个细胞的基因数量直接由糖皮质激素估计有10 - 100,但许多基因是间接监管通过交互与其他转录因子和辅活化因子。GR为绑定到GRE网站corticosteroid-responsive基因的启动子区域。交互的GR二聚体GRE通常增加转录激活。GRs可能增加转录共激活剂分子相互作用,如CBP pCAF,从而激活组蛋白乙酰化和基因转录。例如,相对高浓度的糖皮质激素分泌的增加antiprotease分泌leukoprotease抑制剂(SLPI)上皮细胞14。
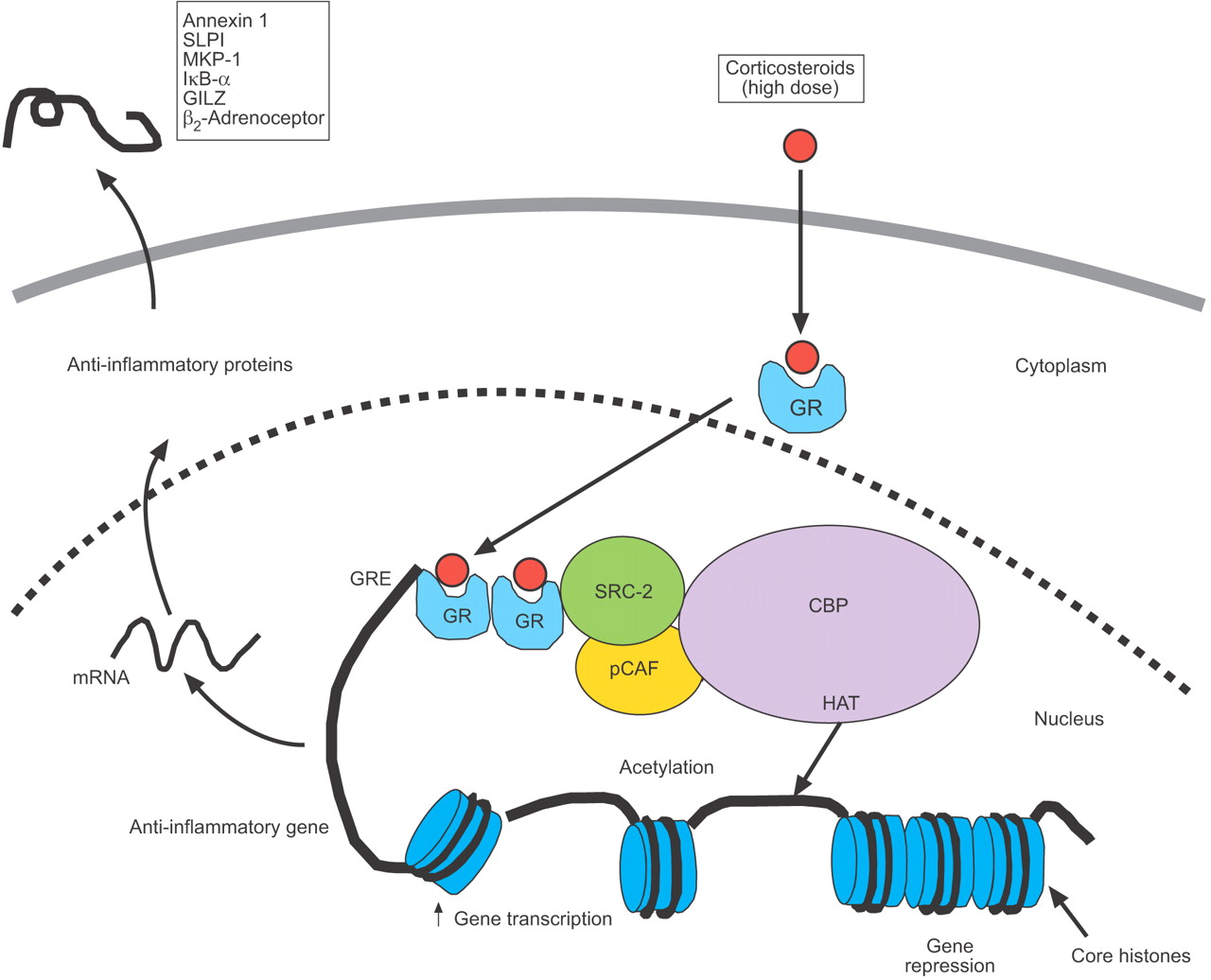
基因的激活糖皮质激素与选择性赖氨酸残基的乙酰化5和16组蛋白H4,导致基因转录增加(图。2⇓)14,28。激活GRs可能绑定到共激活剂分子,如CBP pCAF,以及类固醇受体共激活剂(SRC)和糖皮质激素receptor-interacting蛋白1 (GRIP1或SRC-2),所有拥有帽子活动29日,30.。GRs优先与GRIP1 / SRC-2,随后新兵pCAF31日。
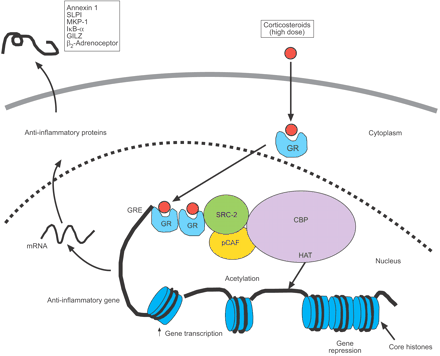
皮质类固醇抗炎基因表达的激活。糖皮质激素与细胞质糖皮质激素受体结合(GRs)把原子核,它们绑定到糖皮质激素反应元素(格蕾丝)steroid-sensitive基因的启动子区域,也直接或间接地共激活剂分子如cAMP-response-element-binding-protein-binding蛋白质(CBP), p300 / CBP-associated因素(pCAF)或类固醇受体共激活剂(SRC) 2,具有内在的组蛋白乙酰转移酶(HAT)活动,引起的赖氨酸乙酰化组蛋白H4,导致激活的基因编码抗炎蛋白,如分泌leukoprotease抑制剂(SLPI),增殖蛋白激酶磷酸酶(MKP) 1、核factor-κB抑制剂(IκB-α)和激素性亮氨酸拉链蛋白质(GILZ)。↑:增加。
抗炎基因激活
几个基因的开启由糖皮质激素抗炎效果,包括膜联蛋白1 (lipocortin-1) SLPI, il - 10的抑制剂NF-κB (IκB-α)。然而,治疗剂量吸入糖皮质激素浓度没有增加膜联蛋白1所示在支气管肺泡灌洗液32,增加IκB-α水平没有被显示在大多数细胞类型,包括上皮细胞33,34。糖皮质激素同时打开两种蛋白质的合成,影响炎症信号转导途径,激素性亮氨酸拉链蛋白质,抑制NF-κB和AP-135和增殖作用(地图)激酶磷酸酶(MKP) 1、抑制p38激酶地图36。然而,似乎不太可能,广泛的糖皮质激素的抗炎作用可以完全解释为增加少量抗炎基因的转录,特别是高浓度的糖皮质激素通常需要这种效果,然而,在临床实践中,在低浓度时糖皮质激素能够抑制炎症。
关掉炎性基因
在控制炎症,糖皮质激素的主要作用是抑制多个炎症通过抑制蛋白质的合成的基因编码(表1⇓)。虽然这最初被认为是通过互动GRs - GRE网站,这些已经被证明是只有少数基因,不包括那些编码炎症蛋白质26。
与转录因子
激活GRs已被证明与其他激活转录因子功能进行交互。大多数哮喘炎症基因被激活的没有GRE网站在基因的启动子区域,然而由糖皮质激素有说服力地压抑。有证据表明糖皮质激素抑制促炎转录因子的影响,如AP-1 NF-κB,调节基因的表达,代码对于许多炎症蛋白质,如细胞因子、炎症酶,粘附分子和炎症受体38,39。激活GRs可以直接与其他交互激活转录因子的蛋白质绑定,但这可能是一个特定功能的细胞中,这些基因是人为的过表达,而不是一个正常细胞的性质。治疗哮喘患者吸入高剂量的糖皮质激素抑制气道炎症不与任何减少NF-κB绑定相关的DNA,但能够关掉炎性基因,由NF-κB gm - csf等40。这表明,糖皮质激素更可能是代理绑定的下游炎性DNA转录因子,和注意力已集中在对染色质结构和组蛋白乙酰化作用的影响。
对组蛋白乙酰化作用的影响
镇压的基因发生逆转的开关炎症基因的组蛋白乙酰化作用41。激活GRs可以直接绑定到CBP或其他辅活化因子抑制他们的帽子的活动14的解除,从而扭转核心组织蛋白的DNA,从而抑制炎症基因。更重要的是,特别是在低浓度可能相关的治疗在哮喘治疗中,激活GR新兵HDAC2激活转录复杂,导致组蛋白脱乙酰作用,,因此,减少炎症基因转录(图3所示⇓)14。使用染色质免疫沉淀反应试验,表明,糖皮质激素相关的乙酰化组蛋白H4招募HDAC2 gm - csf启动子14。通过RNA干扰来选择性地抑制HDAC2上皮细胞系,它已经表明,gm - csf的表达的增加和减少对糖皮质激素的敏感性42。相比之下,混战的HDAC1类固醇响应HDAC3没有这样的效果。尚未解决的一个重要的问题就是为什么糖皮质激素有选择地切断炎性基因,而不影响基因调节扩散,新陈代谢和生存。GRs很可能只绑定到辅活化因子促炎症激活的转录因子,如NF-κB和AP-1,尽管尚不清楚这个特定的识别是如何产生的。AP-1 NF-κB镇压是正常小鼠的GR表达形式不dimerise(渺茫−−/),这表明GR单体能够调节糖皮质激素的抗炎作用,而二聚需要基因活化反应37,43。
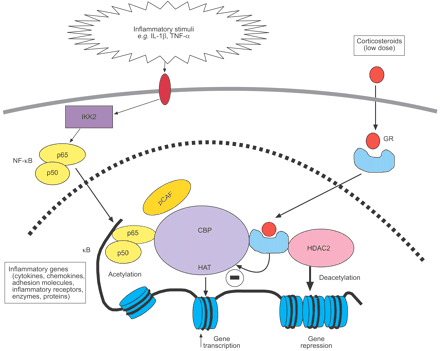
皮质类固醇抑制炎症基因的激活。炎症基因被激活的炎症刺激,如白介素(IL) 1β或肿瘤坏死因子(TNF) -α,导致激活I-κB激酶的抑制剂(IKK) 2,它激活转录因子核转录因子(NF) -κB。p50二聚体和p65 NF-κB把原子核和绑定到特定的κB识别网站并辅活化因子,如cAMP-response-element-binding-protein-binding蛋白质(CBP)或p300 / CBP-associated因素(pCAF),具有内在的组蛋白乙酰转移酶(HAT)活动。这导致乙酰化组蛋白H4核心,导致增加多个炎症蛋白质编码基因的表达。糖皮质激素受体(GRs)、活化的糖皮质激素后,把细胞核和绑定到辅活化因子以抑制帽子活动直接和招聘组蛋白脱乙酰酶(HDAC) 2,改变组蛋白乙酰化作用,导致这些激活炎症基因的抑制。↑:增加;-:抑制。
其他组蛋白修饰
现在已经变得明显,组蛋白核心可能修改不仅通过乙酰化作用,而且通过甲基化,磷酸化和泛素化,这些修改也可以调节基因的转录44,45。组蛋白的甲基化,特别是组蛋白H3,通常由组蛋白甲基转移酶基因抑制的结果46。糖皮质激素的抗炎作用是减少甲基转移酶抑制剂,5-aza-2′脱氧胞苷,表明这可能是一个额外的机制糖皮质激素抑制基因47。的确,之间可能存在交互乙酰化,甲基化和组蛋白的磷酸化,染色质的顺序修改代码(所谓的组蛋白)可能会给特定基因的表达特异性48- - - - - -50。
Nontranscriptional效果
虽然大部分的糖皮质激素介导的转录变化的行为通过染色质重塑,越来越认识到,他们也会影响蛋白质合成减少稳定性的信使rna,蛋白质合成减少。越来越认可的是,一些炎性蛋白mRNA转录水平的调节的稳定性51。这可能是一个重要的抗炎机制糖皮质激素可以关掉后进行生产的炎症蛋白炎症基因被激活。一些炎症基因的稳定性是由监管adenine-uracil (AU)丰富的元素(3′)翻译区域的基因与几个具有约束力的蛋白质,如胡锦涛抗原R(虚)和tristetraprolin,可能稳定mRNA52,53。等炎症基因的基因编码gm - csf和环氧合酶(COX) 2,产生mRNA,尤其容易受到核糖核酸酶的作用,分解信使rna,蛋白质合成因此关闭。糖皮质激素可能抑制影响稳定mRNA的蛋白质,导致更多的快速分解,因此,减少炎性蛋白表达54- - - - - -56。糖皮质激素似乎没有任何影响户珥或tristeraprolin表情,然而57。
对信号转导途径的影响
糖皮质激素信号转导途径上有复杂的影响反式镇压的关键酶参与炎症级联,或通过增加转录这些通路的内源性抑制剂。
增殖蛋白激酶通路
MAP激酶发挥重要作用在炎症基因表达炎性转录因子的调控58。有越来越多的证据表明,糖皮质激素可能对这些通路产生抑制作用。糖皮质激素可以抑制AP-1和NF-κB通过的抑制作用c-Jun n端激酶(物),激活这些转录因子59,60。糖皮质激素减少对一些炎症基因mRNA的稳定性,如cox - 2,通过另一个MAP激酶抑制作用,p38激酶地图53,61年。p38 MAP激酶调节多个炎症基因,包括TNF-αIL-1β,il - 6, gm - csf和引发,而网站在其3′翻译区域,通过稳定他们的信使rna,炎性蛋白的合成增加53。糖皮质激素是介导的抑制作用通过的快速诱导的内源性p38 MAP激酶的抑制剂,MKP-1,基因的开启由糖皮质激素(图4所示⇓)36,62年。在基因微阵列研究中,MKP-1是其中一个最著名的基因激活糖皮质激素63年。糖皮质激素不仅诱导MKP-1基因也减少其退化64年。MKP-1抑制所有MAP激酶通路,因此抑制物,以及在较小程度上,细胞外signal-regulated激酶,除了p38激酶地图62年。这表明糖皮质激素能够抑制所有MAP激酶通路,但MKP-1不同的选择性映射激酶似乎从细胞到细胞65年。皮质类固醇的影响加大了MKP-1表达低浓度氟替卡松加沙美特罗和formoterol66年。这可能有助于增强抗炎作用的糖皮质激素引起的长效β2受体激动剂联合治疗。至少10个其他MAP激酶磷酸酶已经被确认,不同细胞分布和选择性,但尚不确定是否由糖皮质激素诱导67年。
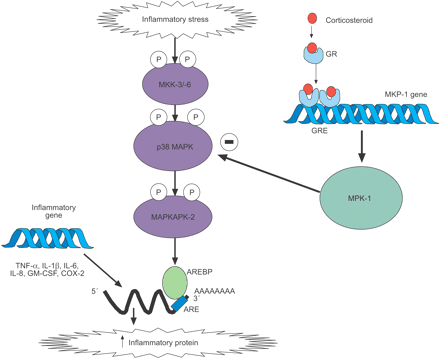
p38增殖抑制蛋白激酶(MAPK)糖皮质激素。p38 MAPK激活炎症强调尽管激活MAPK激酶(MKK) 3和6。p38磷酸化(P) MAPK-activated蛋白激酶(MAPKAPK) 2、在稳定信使rna编码中扮演重要角色的一些炎症蛋白质,如肿瘤坏死因子(TNF) -α,白介素(IL) 1β,IL - 6,引发,集落刺激因子(gm - csf)和环氧合酶(COX) 2。这个信使rna的特点是adenine-uracil-rich元素(战神)3′未翻译区,使mRNA不稳定和快速退化。具有约束力的蛋白质(AREBPs)稳定这些蛋白质,可能被MAPKAPK-2激活(可能是间接的)。糖皮质激素诱导的表达MAPK磷酸酶(MKP) 1、抑制p38和,因此,防止多个炎症蛋白质的稳定。格:糖皮质激素受体;GRE:糖皮质激素响应元素。↑:增加;-:抑制。
皮质类固醇的阻力
虽然糖皮质激素是高度有效的控制哮喘和其他慢性炎症或免疫疾病,一小部分哮喘患者甚至无法应对高剂量的口服糖皮质激素68年- - - - - -70年,COPD患者主要是糖皮质激素反应迟钝71年。抗糖皮质激素的疗效也承认nonpulmonary炎症和免疫疾病,包括类风湿性关节炎和炎症性肠病。Corticosteroid-resistant患者存在相当大的管理问题是很少有另类的抗炎治疗。新见解,糖皮质激素抑制慢性炎症的机制阐明分子基础的皮质类固醇抗哮喘和慢性阻塞性肺病。
Steroid-resistant哮喘
可能会有一些分子机制的抗糖皮质激素的影响,这些病人之间可能有所不同1,69年,70年。很可能有类固醇的光谱响应,非常罕见的电阻一端,相对抵抗的患者需要高剂量的吸入和口服类固醇(激素依赖性哮喘)。
p38增殖蛋白激酶
活检研究展示了典型的嗜酸性粒细胞的炎症反应中发现的这类病人的支气管粘膜,增加2型辅助细胞的细胞因子的表达72年。还有抗糖皮质激素的抗炎作用循环单核细胞73年- - - - - -75年。某些细胞因子(尤其是2、il - 4和IL-13显示表达增加支气管活检标本抗类固醇哮喘反应患者)可能诱发的亲和力GRs的减少炎症细胞如淋巴细胞),导致当地抗糖皮质激素的抗炎作用68年,76年。的组合和il - 4 - 2诱导类固醇阻力在体外通过激活p38 MAP激酶磷酸化GRs细胞核内,减少类固醇亲和力77年。治疗意味着地图p38激酶抑制剂在临床开发可能扭转这种形式的类固醇阻力。
糖皮质激素受体β
另一个提议在哮喘是类固醇抵抗机制GRβ的表达增加,理论上可以作为抑制剂与GRα竞争绑定到GRE网站或共激活剂分子相互作用78年。然而,并没有增加表达GRβ患者单核细胞的激素依赖性哮喘,这显示了在糖皮质激素响应能力在体外。此外,GRα大大胜过了GRβ,使它不太可能有任何功能的抑制作用79年,80年,GRβ察觉哮喘病人的血液中单核细胞81年。此外,没有证据的感应GRβ/ il - 4 - 2暴露,导致皮质类固醇抗单核细胞,令人信服地证明GRβ不能占皮质类固醇抗哮喘81年。
与转录因子
另一个拟议的机制是GRs未能抑制炎症基因的转录因子的激活NF-κB和AP-1等。的确,有缺陷抑制AP-1回应皮质类固醇抗类固醇病人反应的单核细胞75年。这可能是由于增加的激活AP-1由于过度激活物的途径,已证明抗类固醇哮喘病人反应的细胞82年。
有缺陷的组蛋白乙酰化作用
防哮喘病人单核细胞激素依赖性或显示减少抑制细胞因子释放和减少组蛋白H4乙酰化作用在治疗后的核与地塞米松的高浓度(1μM)83年。在一组患者中,核本地化GRs针对高浓度的糖皮质激素是受损,这占了组蛋白乙酰化作用,因为有直接关联的组蛋白乙酰化程度和GR核本地化83年。这可能是由于GR亚硝基化,导致GR从热休克蛋白90的离解84年。然而,在另一组患者中,组蛋白乙酰化作用的缺陷发现尽管GRs的正常核本地化。这可能是由于原子核内GR磷酸化由于p38 MAP激酶的活化77年,这可能会导致失败的招聘不同的共激活剂(年代)。这可能导致GRs的失败反式激活steroid-responsive基因85年。在这组患者中,特定的组蛋白H4赖氨酸乙酰化5由糖皮质激素是有缺陷的83年。这可能意味着糖皮质激素不能激活特定基因的关键高剂量的糖皮质激素的抗炎作用,但是否这是一种罕见的遗传性缺陷尚不得而知。
皮质类固醇的阻力在慢性阻塞性肺病
虽然在哮喘吸入型皮质类固醇激素是非常有效的,它们提供了相对较少的治疗慢性阻塞性肺病的好处,尽管活跃气道和肺部炎症存在。这可能反映了这样的事实:在慢性阻塞性肺病不是由糖皮质激素抑制炎症,无炎性细胞数量减少,细胞因子或蛋白酶水平诱导痰即使口服糖皮质激素89年- - - - - -91年。此外,组织学分析周边航空严重慢性阻塞性肺病患者显示了强烈的炎症反应,尽管用大剂量吸入糖皮质激素治疗92年。有越来越多的证据,一个活跃的类固醇阻力机制在慢性阻塞性肺病,由于糖皮质激素不能抑制细胞因子(如引发和TNF-α),他们通常抑制89年,90年。在体外研究表明,细胞因子释放肺泡巨噬细胞明显抗糖皮质激素的抗炎作用,从细胞从正常的吸烟者相比,这些,反过来,更耐药比不吸烟者肺泡巨噬细胞93年,94年。缺乏对皮质类固醇可能会解释说,至少在某种程度上,吸烟和氧化应激的抑制作用对HDAC函数,从而干扰糖皮质激素的重要的抗炎作用95年。事实上,有HDAC活性之间的相关性和皮质类固醇在细胞因子释放的抑制作用。很可能氧化,在慢性阻塞性肺病特别损害HDAC2 nitrative压力96年阻力,导致皮质类固醇(图5所示⇓)3。虽然这是在慢性阻塞性肺病的各个阶段,它是患者最显著最严重的疾病97年。即使在慢性阻塞性肺病患者已经停止吸烟,类固醇阻力依然存在89年,90年,这些病人已知经验继续氧化应激98年。
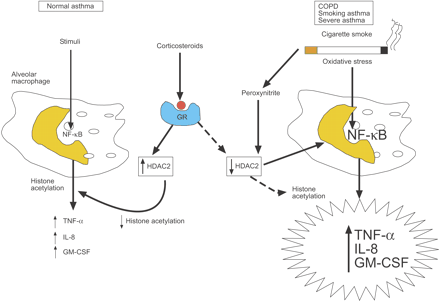
拟议机制的皮质类固醇阻力在慢性阻塞性肺病(COPD),吸烟严重哮喘和哮喘。刺激正常和哮喘肺泡巨噬细胞激活核转录因子(NF) -κB开关组蛋白乙酰转移酶和其他转录因子,导致组蛋白乙酰化作用,随后,转录的基因编码炎症蛋白质,如肿瘤坏死因子(TNF) -α,白介素8 (IL)和集落刺激因子(gm - csf)。糖皮质激素反向通过绑定到糖皮质激素受体(GRs)和招聘组蛋白脱乙酰酶(HDAC) 2。这颠倒了组蛋白乙酰化作用引起NF-κB和开关激活炎症基因。在COPD和哮喘病人吸烟,香烟的烟雾产生氧化应激(代理通过过氧硝酸盐的形成)损害HDAC2的活动。这个放大NF-κB激活的炎症反应,但也减少了糖皮质激素的抗炎效应,因为HDAC2现在无法扭转组蛋白乙酰化作用。类似的机制,严重哮喘可能运行中生成氧化应激增加气道炎症。↑:增加;↓:减少。
氧化应激也增加严重哮喘患者在发作99年- - - - - -101年,这样减少HDAC也占减少糖皮质激素在这些患者和响应能力的哮喘急性加重糖皮质激素相对没有反应。
治疗的影响
现在吸入糖皮质激素作为一线治疗持续性哮喘的治疗成人和儿童在许多国家,因为它们是最有效的治疗哮喘目前可用的102年。然而,在高剂量,系统性吸收的吸入糖皮质激素可能会产生不良的影响,所以一直在寻找更安全的类固醇吸入甚至口服药。
分离糖皮质激素
当前可用的从肺部吸入糖皮质激素被吸收进入体循环,和,因此,不可避免地有一些系统组件。了解糖皮质激素作用的分子机制导致了新一代的糖皮质激素的发展。在发展中这些药物的主要任务是分离的抗炎作用与副作用相关的内分泌动作。正如上面所讨论的,一个主要的糖皮质激素的抗炎作用机制似乎抑制促炎转录因子的影响,如NF-κB和AP-1,激活促炎细胞因子(反式镇压)通过组蛋白乙酰化的抑制作用和组蛋白脱乙酰作用的刺激,没有DNA结合。相比之下,类固醇的内分泌和代谢影响负责系统性皮质类固醇的副作用可能是主要介导的通过通过交互的GRs -格蕾丝(DNA结合独联体镇压)。这导致了糖皮质激素,有选择地寻找小说反式抑制不显著反式激活或独联体镇压,从而减少系统性副作用的潜在风险。由于糖皮质激素绑定到相同的GR,起初这似乎是一个不太可能的可能性,但是,虽然DNA结合涉及GR为,与转录因子的交互AP-1 NF-κB和辅活化因子仅涉及单个GR28。分离反式激活和反式镇压已经证明使用报告基因结构和选择性GR在转染细胞的突变103年。此外,与不dimerise GRs老鼠,没有反式激活,但反式镇压似乎是正常的37,43。此外,一些类固醇,如拮抗剂RU486,表现出更大的反式镇压比反式激活的影响。实际上,今天哮喘中使用的局部类固醇治疗,如丙酸和布地奈德,显示更有效反式镇压比反式激活效应,这也许可以解释他们的选择有效的抗炎剂104年,105年。最近,一种新型类类固醇被描述,有强大的反式镇压与相对较少反式激活。这些分离类固醇,包括RU24858 RU40066,有抗炎作用在体外106年,尽管没有分离的抗炎作用和系统性副作用在活的有机体内107年。其他分离糖皮质激素显示离解在活的有机体内108年。现在几个分离糖皮质激素在临床开发和可能导致吸入类固醇更安全,或者口服类固醇不太可能产生重大不利影响。最近的分辨率晶体结构的配体结合域的GRs可能有助于更好的设计的类固醇109年。
非甾体类抗炎治疗
如今皮质类固醇作用的分子机制已被阐明,这就提出了一个新颖的非甾体类抗炎治疗可能开发模拟炎症基因调控糖皮质激素的行为。许多抗炎作用的糖皮质激素介导的通过抑制转录的影响NF-κB, low-molecular-mass IKK2抑制剂(抑制剂I-κB kinase-2)激活NF-κB现在在临床前开发110年。然而,皮质类固醇的影响除了抑制NF-κB-regulated基因,所以现在还不确定是否IKK2抑制剂将平行糖皮质激素的临床有效性,和他们可能有副作用,如增加对感染的易感性。
正如上面所讨论的,皮质类固醇的疗效是介导通过p38 MAP激酶的抑制,这种激酶也参与了某些情况下的类固醇抗哮喘。现在几个地图p38激酶抑制剂在临床开发和这些药物可能会模仿一些糖皮质激素的影响111年,112年的特定值,但可能是哮喘患者皮质类固醇阻力。
皮质类固醇的逆转耐药性
在慢性阻塞性肺病和严重的哮喘和哮喘病人吸烟,穷人响应性糖皮质激素可能反映了减少HDAC2活动,正如上面所讨论的。茶碱代表第一药物已被证明激活HDAC,导致显著的增强作用的糖皮质激素的抗炎作用113年,114年。这个动作的茶碱不是介导通过磷酸二酯酶抑制或腺苷受体对抗,因此,小说似乎是一个行动的药物113年。在慢性阻塞性肺病巨噬细胞,低浓度的茶碱HDAC活性可以恢复正常和反向这些细胞的抗类固醇在体外114年。临床研究探讨这种影响茶碱正在进行中。它可能会发现其他药物在这门课上,可以形成一个新类的基础抗炎药没有副作用,限制使用茶碱115年。
由于氧化应激和过氧亚硝基似乎在HDAC抑制HDAC活性并模仿缺陷在COPD患者中,抗氧化剂或诱导一氧化氮合酶抑制剂可能是有效的。在发展新的和更有效的抗氧化剂116年和选择性诱导一氧化氮合酶抑制剂已经在临床试验中117年。
结论
糖皮质激素发挥其抗炎作用通过影响多种信号转导途径。他们最重要的行动是关闭多个激活炎症基因通过抑制组蛋白乙酰转移酶和组蛋白脱乙酰酶2的招聘活动炎症基因转录复杂。此外,糖皮质激素可能激活几个抗炎基因和增加某些炎症信使rna编码蛋白质的降解。这一系列的行动可能占的惊人疗效糖皮质激素在复杂的炎症性疾病,如哮喘和很难找到替代消炎药。现在有一个更好的理解如何响应性糖皮质激素减少严重的哮喘,哮喘病人吸烟和慢性阻塞性肺病患者。现在新兴的一个重要机制是减少组蛋白脱乙酰酶2活动由于氧化应激。这些新见解皮质类固醇的行动可能会导致新方法治疗炎症性肺疾病,特别是增加类固醇的效果的情况下,他们不太有效。
脚注
本系列之前的文章:没有。1:风扇J,海勒NM, Gorospe M, Atasoy U, Stellato c .角色趋化因子基因表达的转录后调控炎症和过敏。欧元和J2005;26日:933 - 947。2号:郭Georas SN, J·德·范尼U, Casolaro诉辅助细胞在过敏性疾病2型监管。欧元和J2005;26日:1119 - 1137。3号:Boxall C,霍尔盖特圣,戴维斯DE。转化生长因子的贡献-&β&和表皮生长因子信号在慢性哮喘气道重塑。欧元和J2006;27日:208 - 229。
- 收到了2004年11月1日。
- 接受2005年2月21日。
- ©人期刊有限公司










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}