





文摘
吸入糖皮质激素抑制气道炎症和气道重塑的组件在支气管哮喘。在气管支气管的气道血管,其中包括抑制炎症hyperperfusion,的研究、粘膜水肿的形成,和新血管的形成(血管)。
糖皮质激素目前已知施加其影响气道血管通过基因组和nongenomic机制。基因组行动涉及目标基因的调控,抑制大部分血管气道炎症和血管生成的元素。相比之下,nongenomic行为是由快速的细胞机制,和气道引起短暂的血管收缩,从而扭转炎症hyperperfusion。
糖皮质激素导致的血管行为控制哮喘的临床症状主要是通过影响呼吸道口径的肺外围和气道代答。
综述文章,最新进展的了解细胞机制和临床意义的交互的吸入型皮质类固醇激素和哮喘气道血管进行了综述。
支气管哮喘的气道炎症是一个核心特征。除了在支气管壁炎性细胞浸润1,组织学分析支气管活检标本和血液流动的新方法测量显示突出的改变支气管哮喘患者(气管)脉管系统。主要的结构和功能变化与气道循环包括血管(血管)的扩散2- - - - - -4,增加血流量5,6、微血管通透性增加7,8和水肿形成气道壁9。这些血管组件似乎有重要的临床意义,因为他们的哮喘严重程度的相关性,包括气流限制10- - - - - -13和支气管高反应性7,11,14- - - - - -18。最新进展的了解细胞机制负责这些血管异常可能最终导致新的治疗方法治疗哮喘。
自1949年以来,当Henchet al。19他的发现发表在可的松造成戏剧性的改善类风湿性关节炎患者,糖皮质激素已成为建立最有效的抗炎药物药物治疗各种慢性炎性疾病,包括哮喘20.。在哮喘炎症过程涉及到各种炎性趋化因子的表达增加,细胞因子,生长因子,脂质介质,粘附分子,酶和受体相同的炎症介质21。糖皮质激素是最有效的药物来抑制气道炎症,炎性的差别主要是通过对这些基因的蛋白质22,23。此外,糖皮质激素似乎asthma-induced的反向组件结构变化(气道重塑),包括增加支气管壁的血管24。
糖皮质激素的抗炎作用发生的相当大的延迟(在几小时或几天),因为所需的细胞行为的多个步骤改变蛋白表达。然而,现在证据积累的快速皮质类固醇的行动25,26膜结合类固醇受体的存在,可能调解这些快速行动27,28。糖皮质激素的快速效应也被证明在气道近年来血管29日,30.。这些“模”行动可能导致糖皮质激素的抗炎作用;然而,潜在的细胞机制显然是与转录调控的不兼容(基因组)的行动。
这里,快速扩张的当前作者概述信息:1)基因组的分子基础和糖皮质激素的nongenomic行动;2)哮喘的血管表现;和3)交互的皮质类固醇和气道血管炎性改变的哮喘患者的气道血管可以逆转。
皮质类固醇的行为基因组机制
吸入糖皮质激素抑制气道炎症,负责asthma-associated气道血管的变化。糖皮质激素的抗炎效应是由于目标基因的活化或抑制炎症过程(图1所示⇓)。这些基因组行动是由细胞内受体(糖皮质激素受体;GRs),最终改变通过直接DNA结合转录31日或转录因子失活32。因为基因组机制需要额外的步骤,如蛋白质合成、整理、修改、和胞内运输,抗炎作用的糖皮质激素至少需要几个小时发生。
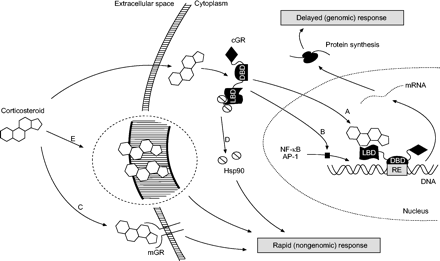
原理图的复杂的糖皮质激素细胞的行为。基因组行动是由胞质受体,最终通过改变转录)直接DNA结合转录因子失活或B)。相比之下,nongenomic行为是由C)膜结合或D)胞质受体,或E)非特异性与细胞膜的相互作用。cGR:细胞质糖皮质激素受体;经理:膜糖皮质激素受体;小黑裙:配体结合域;DBD: dna结合域;一半:热休克蛋白90;再保险:响应元件;NF-κB:核factor-κB; AP-1: activating protein-1.
基因活化直接DNA结合(transactivation)
根据甾类激素作用的经典模型,亲脂性的激素分子很容易穿过细胞膜的脂质双分子层进入细胞和绑定到特定的受体33。皮质类固醇绑定和离解后两个单元的热休克蛋白90(一半),以作为分子伴侣蛋白,激活GRs把原子核和绑定到复发的DNA序列(响应元素)在目标基因的启动子区域。根据监管的积极或消极的活动元素,绑定ligand-activated受体的复杂的可能,或表达下调基因转录,因此蛋白质合成。糖皮质激素增加一些抗炎基因的转录通过直接DNA结合;然而,这种机制似乎有轻微的炎症的抑制作用。例如,糖皮质激素有效抑制炎症已报告有缺陷的小鼠GR,不能结合的DNA34。最近,它已建议transactivation负责一些副作用(如。糖尿病感应,糖皮质激素引起的皮肤萎缩)35。
快速、NONGENOMIC皮质类固醇的行为机制
虽然主要的抗炎作用的糖皮质激素是由于转录机制,越来越多的证据也表明行为体现在秒或分钟。这些影响是由细胞机制太快速涉及基因表达和被称为nongenomic行动43。Nongenomic行动是由特定的相互作用与膜结合或细胞质GRs或非特异性与细胞膜的相互作用(图。1⇑)。尽管高机械的多样性在不同的细胞类型,nongenomic行动有一些共同的特征(表1⇓)。
与基因组行动,更了解快速nongenomic行动糖皮质激素的临床意义。在支气管哮喘,吸入丙酸已被证明能够敏锐地减少气道粘膜血流量29日,44,抑制吸入AMP-induced支气管收缩45,防止运动性儿童支气管阻塞46。糖皮质激素也被证明在过敏性鼻炎严重减少鼻痒47。符合的早期报道急性支气管扩张剂的强化行动皮质类固醇48,49,布地奈德敏锐地增强的快速出现支气管扩张剂行动formoterol慢性阻塞性肺疾病患者在最近的一项研究50。尽管快速影响气流限制也被建议在急性哮喘患者51,没有足够的证据表明吸入激素治疗肺功能结果在临床上重要的快速变化52,而系统性皮质类固醇可能要求> 6小时24小时改善肺功能53。
膜receptor-initiated行动
糖皮质激素诱导的细胞快速没有进入细胞的影响。在某些情况下,这些细胞membrane-initiated行动也敏感的细胞质GR的阻滞剂。这些观察最终导致皮质类固醇结合位点的发现,与细胞膜受体相关。早期的演示后的高亲和性细胞膜结合位点在大鼠肝脏糖皮质激素54所示,膜结合受体两栖动物的大脑55和小鼠淋巴瘤细胞56。膜结合GRs也存在于各种各样的人类细胞,包括进展期57和外周血单核细胞28。最近,皮质类固醇的膜结合位点已被证明在气道平滑肌细胞分离人类血管58,59。这个膜受体配体结合可能负责激素对去甲肾上腺素的抑制作用摄取到这些细胞60。
膜的分子结构GR仍然是未知的。受体分布的分析表明,相同的受体可能在两种核函数和膜的位置27。膜受体是一种改性的神经元细胞的古典GR在某些情况下61年。相反,过度的His-tagged胞质受体没有增加膜受体的表达在最近的一项研究28,这表明这些都不是可互换的受体数量。支持不同的受体蛋白的存在,两栖动物的大脑神经元膜受体已经被确认为一种酸性糖蛋白的表观分子量63 kD62年,这是不符合的特点和预测体重(即。96 kD)的胞质GR蛋白质。膜受体激活已被证明诱导快速影响多种第二信使系统(Ca2 +,腺苷3′,5′一磷酸、肌醇联结,蛋白激酶C)改变细胞过程63年,64年。除了膜相关受体,糖皮质激素可以绑定其他受体,离子通道、酶、转运蛋白、细胞膜之前从未描述的蛋白质27。
细胞内receptor-initiated行动
激活的细胞质GRs已被证明发起快速nongenomic行动。配体结合后,热休克蛋白(如。一半)和增殖蛋白激酶的激酶系统(如。Src)正在迅速释放GR-chaperone蛋白质复杂,和释放的蛋白质被认为引起快速的细胞反应65年。例如,热休克蛋白似乎调解dexamethasone-induced钙调磷酸酶活性增加肾导管66年已报告,而Src蛋白质负责dexamethasone-induced抑制磷脂酶A2在肺腺癌的细胞67年。配体结合的胞质GR也会导致快速激活的内皮细胞一氧化氮(NO)合成酶;急性药理剂量的管理机制(40 mg·公斤−1)地塞米松似乎减少血管炎症和降低心肌梗塞大小动脉缺血和再灌注损伤后小鼠68年。
非特异性的行动
糖皮质激素可能引起快速的影响通过改变细胞膜的物理化学性质。亲脂性的激素分子插入到细胞膜的磷脂影响。在高浓度时,糖皮质激素往往积累膜,膜流动性的变化,从而引起细胞功能快速变化69年。由于膜流动性的影响需要皮质类固醇浓度> 10−4米,这种机制功能的重要性也很成问题。皮质类固醇夹层在细胞膜中,然而,可能会影响整体的功能蛋白(即。离子通道、转运蛋白和受体)远低于这个浓度范围61年。例如,糖皮质激素改变膜运输阳离子膜夹层和增加线粒体质子漏的70年。在人类支气管动脉平滑肌细胞分离,糖皮质激素强烈抑制有机阳离子的吸收(或extraneuronal吸收)60;这是去甲肾上腺素失活在航空公司的关键71年。这种快速nongenomic行动可能会增加去甲肾上腺素/血管收缩神经信号的持续时间,从而减少气道血流如健康和哮喘吸入糖皮质激素后29日。类似的现象,抑制去甲肾上腺素吸收进大脑的神经胶质细胞,提出了增强去甲肾上腺素的积累在突触,并可能加速去甲肾上腺素再摄取抑制剂的抗抑郁药的临床效果72年。
哮喘患者的气道血管< 1材质? >
除了作为支气管壁的营养来源73年,气道循环热量和水交换的重要角色74年,75年、气道代7,14,17,76年,并可能肺部呼吸道口径外围的监管10- - - - - -13,77年- - - - - -80年。此外,气道循环函数作为炎性细胞的渠道招聘81年,82年,参与当地公布的间隙,生物活性物质83年,84年和吸入材料运输从表面到深层组织85年。
作为呼吸道疾病特征同时连续炎症和修复过程,哮喘是已知的导致功能性(血管舒张,hyperperfusion微血管通透性增加,水肿形成和炎症细胞招聘)和结构变化在气道血管(血管)。血管的变化有显著的病理生理学后果,因此可能参与哮喘的临床表现86年,87年。尽管炎症介质的复杂行为,神经递质和神经肽在血管内皮和平滑肌细胞,细胞机制导致的血管表现哮喘尚未完全阐明。
增加血流量
炎症充血和hyperperfusion是一致的特性。哮喘,因此,预计将增加气道血液流动。在过敏性哮喘的绵羊模型,吸入的挑战与抗原导致气道血流量增加,炎症介质有关88年- - - - - -90年。使用最近发明了无创性气道血流量测量方法,随后这些发现已在人类被试。通过测量吸入的吸收,可溶性惰性气体(二甲醚)91年,92年、气道黏膜血流量(包括∼70%的总气道血液流动)已被证明是稳定的支气管哮喘患者增加5,29日,93年,94年。计算的体积进行航空公司从气管终端细支气管(无视最近50毫升),意思是气道血流值在哮喘患者24 - 77%高于健康对照组5,94年。
在哮喘气道,血流增加是由于扩张的阻力动脉和血管数量的增加。在动物模型中,大多数炎症介质引起血管舒张73年。组胺,最初有三相的影响血管舒张血管收缩,然后一个持久的血管舒张。感觉神经肽释放传入神经也强烈的血管舒张95年。尽管明显的血管舒张,一些介质在哮喘患者的气道血管收缩剂,包括endothelin-196年,似乎负面受促炎症肿瘤坏死factor-α97年。还需要进一步的研究来揭示气道hypervascularity的贡献在哮喘患者气道血流量增加。
异常的血液流动的监管
交感神经系统提供了局部控制气道血流通过释放去甲肾上腺素,引起血管收缩,通过激活α1-adrenoreceptors血管平滑肌98年- - - - - -One hundred.。的α1肾上腺素的反应气道血液流动的证实在活的有机体内另外,哮喘病患者的疗效94年。气道血管高反应性,这也被报道在动物模型的特异反应性99年,类似于α-adrenergic受体激动剂在哮喘患者的支气管高反应性101年。asthma-associated血管高反应性的可能机制包括α-adrenoceptors表达和功能的增加,改变了信号转导,受损α-adrenergic受体激动剂的失活细胞吸收过程,或组合102年,103年。一个假定的内皮收缩因子也被提议负责血管α敏感性增加1肾上腺素能受体激动剂99年。
虽然β2肾上腺素能受体激动剂引起的血管舒张支气管循环主要是通过增加内皮细胞的合成104年,105年,最近的研究披露钝化β2-adrenoreceptor-mediated哮喘病患者的气道血管舒张94年,106年。除了β-adrenergic受体的信号转导或减少受损表现102年,β的衰减2肾上腺素能agonist-induced血管舒张,也许是因为最大血管舒张达到在气道炎症反应5。
增加血管通透性
的研究炎症和水肿形成共同的特征。过敏性哮喘患者,吸入过敏原产生直接和晚期炎症反应期间血浆外渗发生气道微循环107年- - - - - -109年。Asthma-associated等离子体泄漏被认为发生主要通过细胞间差距的形成否则postcapillary小静脉内皮细胞紧密相关110年- - - - - -112年。,等离子体可以通过上皮细胞,收集在航空公司,妥协上皮完整,纤毛的功能,从而导致过度气道分泌物形成腔的粘液栓113年。等离子体泄漏也会导致粘膜水肿和支气管壁肿胀,这可以减少呼吸道口径,导致气流限制80年。此外,微血管通透性的增加可能导致哮喘支气管高反应性8,18。
血浆外渗asthma-associated炎性反应被认为是一个特定的侮辱涉及炎症介质的释放、生长因子、神经肽,嗜酸性粒细胞颗粒蛋白,细胞因子、蛋白酶的气道109年,114年。组胺,platelet-activating因子,白三烯D4缓激肽可以通过细胞间差距的形成微血管通透性增加115年- - - - - -118年。诱导痰的血管内皮生长因子(VEGF)水平,这已被证明导致内皮细胞的外窗119年,增加120年和与哮喘患者的气道血管渗透性8,121年。在啮齿动物与发现122年,123年“神经源性炎症”的现象似乎有次要角色的渗透率增加哮喘因为吸入刺激物辣椒素激活无髓鞘的感觉神经释放炎症神经肽(即。P物质、降钙素相关基因肽)唤起没有等离子体在人类呼吸道的分泌物124年。除了炎性渗出性代理、血管舒张和微血管阻塞微血管通透性增加125年。最近,新生成的血管在慢性气道炎症是漏水的,不成熟,不稳定3,这可能导致血管通透性增加哮喘。
炎症细胞招聘
哮喘的血管表现包括气道微循环白细胞的涌入到血管外的组织81年,82年。招募白细胞是主要由炎症反应释放肽趋化因子126年和脂质介质,如白三烯B4和前列腺素D2127年。细胞粘附分子的特定子集白细胞和内皮细胞负责不同步骤的白细胞外渗128年。白细胞的主要血管轮回的步骤通常被称为白细胞沿着内皮表面滚动,激活炎症化学引诱物,绑定到内皮细胞间粘附分子,溢出,趋化性的炎性组织损伤129年,130年。虽然炎症导致postcapillary小静脉内皮间隙和等离子渗漏,白细胞迁移主要发生在收集小静脉的气道111年。
血管生成和微血管改造
气道循环是已知的在各种病理条件下增殖。除了肺部动脉阻塞131年、肺脓疡132年、肺结核133年、肺肿瘤134年,气道循环已被证明在慢性气道炎症性疾病发生结构性的变化135年。慢性炎症导致的增长从现有的新血管(血管)和改变现有的血管(微血管改造)。在哮喘患者中,早期组织学研究表明,异常厚支气管粘膜包含放大和拥挤的血管136年,137年。自那时以来,许多研究已经证明增加血管质(即。大小和数量的船只,或血管截面积)与哮喘支气管粘膜的专利10- - - - - -13,15- - - - - -17,138年- - - - - -141年。Asthma-associated炎症也会导致血管功能改变的气道异常血液流动的监管29日,94年。
多种生长因子、细胞因子、趋化因子、酶和其他因素(如。缺氧)据报道,诱导血管生成142年,但没有明确各自的角色在哮喘。因为哮喘的气道VEGF水平增加,直接与气道粘膜血管,反向与气道功能和代答7,15,16,120年,VEGF可能的关键调节器哮喘的新血管形成。此外,在lung-targeted转基因老鼠,选择性超表达VEGF诱导的会更加表型与血管重塑(血管生成,研究进展,水肿形成)143年和没有血管的重塑(粘液化生,肌细胞增生)气道高反应的航空公司144年。此外,肿瘤坏死factor-α,把增长factor-αinterleukin-8145年,矩阵改造蛋白酶146年,基质细胞衍生因子- 1140年,基本成纤维细胞生长因子147年,血管生成素16都提出了一些在血管和微血管改造在哮喘中的作用。
气道粘膜的扩大和拥挤的血管导致气道壁厚增加哮喘和,因此,是潜在的捐助者气流限制和气道高反应性80年。气道血管和之间的相关性被发现在哮喘气道高反应11,15,16;是否存在因果关系仍有待证明。
快速的糖皮质激素对气道血管的影响
糖皮质激素发挥快速、延迟和长期效应在哮喘气道血管(图2所示⇓)。在这些影响中,糖皮质激素可以敏锐地(几分钟后)改变血管张力通过nongenomic细胞行为。1962年,麦肯齐和斯托顿是第一个报告,糖皮质激素应用于漂白皮肤原因由于局部血管收缩148年。虽然局部皮质激素治疗后皮肤漂白需要几个小时出现,皮内注射糖皮质激素诱发热烫不到一个小时,这表明nongenomic行动。类似的现象在大鼠坐骨神经最近报道149年和人类支气管粘膜29日。糖皮质激素也能敏锐地加强对血管收缩剂的反应在一些系统性血管床,包括径向150年,冠状动脉151年,眼152年,支气管动脉153年。此外,糖皮质激素可能强烈(60分钟)内恢复血管收缩剂对去甲肾上腺素的反应患者的医疗条件受损的内源性糖皮质激素的生产154年,在实验肾上腺切除术155年。
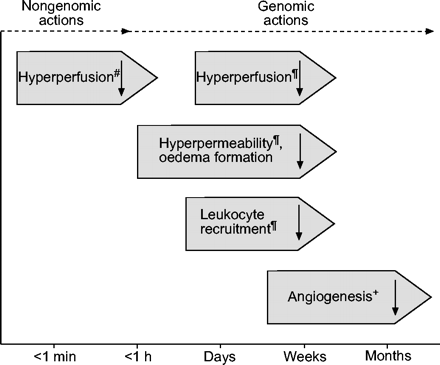
快速(#),延迟(¶)和长期(+)血管影响的吸入型皮质类固醇激素在哮喘患者的气道。影响垂直间隔的y轴上只是为了方便阅读。
减少气道血液流动
吸入糖皮质激素可以敏锐地抑制气道hyperperfusion与哮喘有关。单剂量吸入丙酸已被证明会降低气道粘膜血流量在健康和哮喘受试者最大效应∼30分钟吸入后,并在90分钟回到基线29日。血液流动效果增加剂量依赖性的方式880µg丙酸,有哮喘病患者比健康对照组显著更大的影响。急性血管收缩的动作也被证明在吸入的二丙酸倍氯米松和布地奈德44。
到目前为止,证据表明,糖皮质激素减少气道血流通过调制norepinephrine-mediated(同情)血管张力的控制。特拉唑嗪预处理5毫克,这是一个选择性α1肾上腺素能受体拮抗剂,抑制丙酸对血流的影响153年,这表明糖皮质激素促进去甲信号传输诱导神经源性血管收缩。有许多可能的机制来促进去甲信号传输156年。在原发性高血压、神经受损的去甲肾上腺素提出了负责增加同情的语气157年。相反,糖皮质激素被强烈地抑制去甲肾上腺素的extraneuronal吸收158年,从而增加交感神经信号传输159年和血管的语气160年。气道,糖皮质激素抑制extraneuronal单胺transporter-mediated去甲肾上腺素的支气管动脉平滑肌细胞58,60。这种快速效应降低去甲肾上腺素释放的清除气道神经,因此,可能负责α1-adrenoreceptor-dependent血管收缩剂糖皮质激素的作用(图3所示⇓)。此外,去甲肾上腺素的特异反应性extraneuronal摄取受损,在ovalbumin-sensitised兔子103年,可能导致增加血管收缩剂反应,糖皮质激素如哮喘科目29日。糖皮质激素也将迅速提高阳离子药物的药理作用extraneuronal吸收的灭活161年。支持这个概念发现吸入丙酸敏锐地强化吸入α的血管收缩剂的效果1肾上腺素能受体激动剂153年。
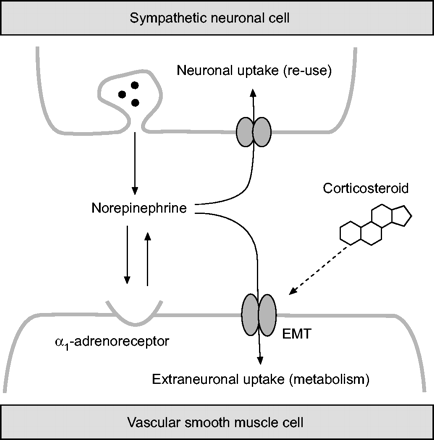
拟议机制的急性呼吸道吸入型皮质类固醇激素的血管收缩的影响。糖皮质激素促进去甲(同情)神经肌肉信号传输通过迅速(5分钟内)抑制extraneuronal单胺转运体(EMT)在血管平滑肌细胞。
另外,糖皮质激素的急性血管效应可能涉及其他nongenomic行动,如磷酸肌醇分解的快速刺激增加细胞内肌醇1,4,5-triphosphate水平162年的蛋白激酶C-dependent激活细胞膜Na+/小时+换热器163年和血管平滑肌细胞的细胞质中的钙动员164年。
其他快速的效果
皮质类固醇可能会扰乱对细胞通过快速抑制炎症过程操作流程或介质与炎症有关65年,165年,166年。例如,糖皮质激素将会快速抑制蛋白水解酶的释放稳定鼠肝脏溶酶体膜167年、表皮生长factor-stimulated磷脂酶A2活动和花生四烯酸释放人类腺癌细胞系A54967年,168年。他们也抑制阳离子骑车穿过细胞膜,呼吸在淋巴细胞有丝分裂原激活的伴刀豆球蛋白A169年在巨噬细胞吞噬作用和超氧化物阴离子生产170年,171年内皮细胞激活和leukocyte-endothelial交互165年外周血中性粒细胞,脱粒172年。的快速抑制炎症反应可以减少asthma-associated的研究,形成水肿,炎性细胞在航空公司招聘;然而,这些严重影响糖皮质激素的不良记录患者的哮喘。
糖皮质激素在气道血管延迟的影响
糖皮质激素与脉管系统维护和交互,超过,增强血管的基调173年。受损的内源性糖皮质激素的生产各种医疗条件(如。急性肾上腺机能不全)通常与全身性低血压有关。相比之下,皮质类固醇管理或生产过剩的内源性糖皮质激素(如。库兴氏综合征)被认为是诱发系统性高血压,部分通过增加外周血管的基调。血管的影响很大程度上是通过基因调控的机制,因此发生的延迟几小时或几天。
在哮喘吸入糖皮质激素可以抵消所有的血管表现气道炎症通过直接作用于血管平滑肌或内皮,或间接通过抑制炎症介质释放血管活性的174年,175年。因为疾病严重程度似乎与哮喘患者气道血管变化,这些基因血管行动的吸入型皮质类固醇激素治疗价值和最近收到可能增加兴趣176年。
减少气道血液流动
大量的研究已经证明,吸入激素治疗哮喘患者的气道粘膜血流量降低。在横断面研究中,气道粘膜血流量高于∼23% corticosteroid-naive corticosteroid-treated哮喘患者5。在介入研究中,吸入丙酸(440µg·天−1)治疗2周血流量减少了∼12%和21%在两个不同的组的哮喘患者6,93年,另外表明气道血液返回到预处理级别2周后停止治疗。
糖皮质激素被认为降低气道粘膜打击流动通过抑制炎症和血管舒张药的释放介质。这是支持的事实,吸入丙酸的影响(440µg·天−1)是类似于白三烯修饰符montelukast (10 mg·天−1),减少气道哮喘患者在治疗2周内血液流动6。糖皮质激素也可能直接作用于血管平滑肌和内皮细胞(表2所示⇓),从而提高血管张力,减少打击气道血管中流动。在内皮细胞,糖皮质激素可以降低有效的可用性血管舒张药没有187年,可能通过抑制内皮没有合酶的活动188年或通过增强没有消除活性氧的生产过剩191年。在血管平滑肌细胞,糖皮质激素增强应对内源性血管收缩剂通过多种机制。等,这些可能包括增加受体表达179年由receptor-G蛋白质跨膜信号耦合192年、钙流入193年蛋白质和钙敏感性的收缩194年。
影响血液流动的监管
最初的报道表明,糖皮质激素可以恢复血管反应肾上腺素能受体激动剂94年,符合观察肾上腺素的支气管平滑肌的响应能力受损。而β2-adrenoreceptor-mediated corticosteroid-naive哮喘患者气道血管舒张是钝化94年,106年吸入沙丁胺醇治疗的血管舒张效应出现在哮喘患者吸入皮质类固醇治疗5。血管舒张反应吸入沙丁胺醇治疗也恢复了吸入丙酸治疗2周(440天µg·−1)93年。目前,没有报告可以在吸入激素治疗对α的影响1肾上腺素血管高反应性哮喘病人。
抑制水肿形成的
糖皮质激素抑制微血管通透性增加,水肿的形成与气道炎症有关。大鼠气管、地塞米松治疗降低血浆外渗介质产生的神经源性炎症,如速激肽和P物质195年。在ovalbumin-sensitised老鼠,地塞米松治疗减少蛋白质外渗应对接触卵白蛋白过敏性炎症引起的118年。哮喘患者吸入激素治疗可以抑制微血管通透性增加,血浆泄漏进入气道腔,由测量浓度的高分子量蛋白(如。α2巨球蛋白)诱导痰121年,196年和支气管肺泡灌洗液14,197年。因为在asthma-associated气流阻塞微血管通透性增加的作用,糖皮质激素的抗渗行为可能的治疗价值。
在神经源性炎症、糖皮质激素的抑制作用可能是由中性肽链内切酶、血管紧张素转换酶由于地塞米松的行为完全逆转通过抑制这些tachykinin-degrading酶198年。血浆蛋白抑制气道外渗,地塞米松似乎块肥大细胞脱颗粒,从而释放白细胞三烯B4和C4ovalbumin-sensitised老鼠的航空公司针对抗原的挑战118年。在最近的一项研究哮喘受试者接受吸入二丙酸倍氯米松(800µg·天−1之间的显著相关性被发现),减少气道血管渗透性和诱导痰液VEGF水平8;这是符合这个概念,但没有明确的证据,吸入糖皮质激素可能降低气道的研究通过减少气道的VEGF水平,一种潜在的刺激血管渗透性。
抑制炎症细胞的招募
白细胞和内皮细胞粘附分子极度参与白细胞从血液外渗到支气管旁周围组织炎症气道疾病,因此,这些分子通常被认为是抗炎治疗干预的目标128年。通过干扰这些招聘的关键分子级联199年从post-capillary小静脉,糖皮质激素抑制白细胞移民到血管外的矩阵,从而减少气道壁的炎症细胞浸润24。众所周知,糖皮质激素干扰白细胞和内皮细胞粘附分子通过多种机制165年;然而,这些都不完全阐明哮喘。基因组行动已被证明减少多种粘附分子的表达(如。细胞粘附分子1和3,E-selectin血管细胞粘附分子1,淋巴细胞抗原关联)涉及招聘在哮喘气道炎症细胞22,199年,200年。糖皮质激素可以抑制细胞因子和趋化因子的产生负责增加粘附分子的表达与炎症相关的条件201年,202年。此外,提出了糖皮质激素诱导的合成抗炎介质(如。膜联蛋白I),抑制细胞粘附分子的功能165年。
糖皮质激素的长期影响气道血管
目前治疗方法抑制气道炎症不能控制所有哮喘的症状。因此,注意力都集中在气道炎症反应的结构性变化(集体称为气道重构)近年来。形态变化与加速呼吸功能恶化,不可逆或部分可逆气流阻塞,和持久一些哮喘患者支气管代答203年,204年。然而,哪些元素导致气流阻塞气道重构的发生和气道代答,这些效应的大小仍受争论。
其他asthma-associated气道粘膜结构改变,定量(血管)和定性(微血管改造)气道血管的变化似乎与疾病严重程度,包括肺功能12,13和支气管代11,16,18。因此,气道血管的形态学改变现在被认为是潜在的治疗目标;然而,他们的角色在哮喘的病理生理学仍然投机。
回归的气道血管
糖皮质激素有潜在的治疗作用的逆转的组件在哮喘气道重塑。这是测试一些最近的研究使用IV型胶原蛋白单克隆抗体识别血管支气管哮喘病人标本获得。横断面研究,与吸入二丙酸倍氯米松治疗已被证明是与固有层占据的面积减少了船;然而,船舶的数量只有在减少病人在高剂量(≥800天µg·−1吸入皮质类固醇)11。基于介入研究,每日吸入的剂量和治疗的长度似乎吸入型皮质类固醇激素的血管效应的关键因素。此外,抑制对改造过程的影响似乎仅发生在糖皮质激素长期治疗。而6个月治疗剂量800µg二丙酸倍氯米松减少血管和血管区域的数量139年六周治疗卡松丙酸只有有效地每天吸入剂量1000µg,而不是200µg,显著降低血管和血管区域的数量17。此外,在哮喘患者接受每日剂量的400 - 1000µg吸入皮质类固醇(二丙酸倍氯米松或布地奈德),添加200µg每日剂量丙酸对气道血管没有显示任何影响205年。
长期的抑制效应与吸入糖皮质激素治疗气道血管生成可能涉及angiogenetic因素与气道炎症的抑制;然而,这些仍需探索。
临床意义
最近的研究表明,吸入糖皮质激素有重要的作用在气道血管张力的调节,渗透率和结构在哮喘患者。这些血管的影响糖皮质激素在气道血管可以预期管理哮喘诊断和治疗的影响176年。
尽管日益升值的基因组和nongenomic机制造成血管的行为,仍有巨大的差距的理解模式的皮质类固醇行为及其对哮喘的病理生理学的影响,包括疾病严重程度和持久性。此外,在哮喘患者没有皮质类固醇治疗反应良好206年,这两个基因的不敏感和nongenomic皮质类固醇的行为还有待建立。
气道炎症的生物标志物血流量
有一个正在进行的搜索的气道炎症标记可以作为索引的独立评估肺功能疾病的严重程度和用于量化反应在哮喘患者抗炎治疗207年。使用了一些侵入性和非侵入性的方法,包括分析细胞和非细胞成分诱导痰及支气管肺泡灌洗液、支气管活检标本的组织学检查,化验血清炎性标志物,呼出气体浓度的测量(如。没有),炎症介质在呼出的气息凝结。随着无创方法测量气道血液流动5,92年、气道hyperperfusion最近提出的生物标志物asthma-associated炎症,鉴于血管hyperperfusion组织炎症的一个一致的特性。报道减少抗炎治疗哮喘患者的气道血液流动,人们支持这个概念6,93年。
组织生物利用度的吸入型皮质类固醇激素
标准的在活的有机体内筛选试验等级相对抗炎的效能和组织局部皮质激素治疗的生物利用度是麦肯齐“皮肤漂白”测试148年。测试是基于糖皮质激素引起相对快速的能力(几小时)局部应用后皮肤血管收缩208年。尽管它良好的相关性与GR结合亲和力209年,麦肯齐测试远非理想预测哮喘吸入糖皮质激素的抗炎的效能。这是在最近的一项研究显示无显著相关性的影响你与吸入布地奈德治疗(400µg·天−1)在支气管metacholine代答,呼出没有水平,血嗜酸性粒细胞计数和布地奈德对麦肯齐测试的影响患者的哮喘210年。
在活的有机体内皮质类固醇的活动是依赖药物吸收的速度和程度,可用网站的行动211年。这可以通过测量测试气道吸入型皮质类固醇激素血流量响应,这似乎是理想的评估这些药物进入他们的网站在支气管壁的行动29日,44。因此,分析快速血管收缩剂效果的吸入型皮质类固醇激素可以提供详细的信息在他们的气道组织生物利用度,这取决于aerosolised药物的物理化学特性和aerosol-generating设备。然而,需要进一步的研究来建立联系的快速nongenomic血管收缩剂和吸入糖皮质激素的抗炎作用。
气道电导
气道血管充血,粘膜下水肿,细胞腔的流体积累都提出了导致过度在哮喘气道狭窄和气道代主题。虽然气道血管被占据很大一部分内支气管壁212年,213年关于气道血液和血管充血报告是相互矛盾的78年,213年- - - - - -215年。较少争议关于气道水肿的影响,缩小了航空公司通过增加内支气管壁的厚度和导致气道代答到相同的程度,气道肌肉缩短会导致更大的腔的狭窄较正常的气道9,216年。
直接吸入糖皮质激素对肺功能的影响似乎是边际急性哮喘患者29日,52,53。被耽搁的抑制性行为在呼吸道充血,研究进展,水肿形成,血管生成表明,长期与吸入糖皮质激素治疗可能会显著降低血管腔室的大小,从而降低气流阻塞。然而,这些发现证实了未来的研究。
与吸入支气管扩张剂的互动
气道循环有重要作用从呼吸道吸入药物的间隙,和系统性药物分布的气道。在哮喘气道炎症增加血流量预计将提高间隙,因此,减少吸入的影响的大小和持续时间bronchoactive药物。相比之下,增加气道血流可以支持航空公司系统管理药物的分布。因此,气道炎症增加血液流动似乎临床有益,以及不良的影响,在哮喘的药物治疗87年。
气道的类固醇诱导性减少血流量可能会提高吸入支气管扩张剂的行动通过减少气道的间隙。虽然尚未直接验证这个假说在哮喘患者中,有证据支持增强化合物的生物效应与血流受损交付给航空公司83年。此外,快速气道吸入布地奈德引起的血管收缩44可能是负责提高吸入支气管扩张剂行动formoterol见慢性阻塞性肺疾病患者50。自从类固醇诱导性血管收缩峰值迅速(∼30分钟后药物吸入),同时政府的吸入型皮质类固醇激素和支气管扩张药可能的临床意义。
结论
糖皮质激素的复杂血管行动表明,asthma-associated血管生成,hyperperfusion,研究进展和白细胞招募抗炎的目标。涉及基因组药物抑制影响行动。最近演示了快速nongenomic糖皮质激素对气道血管平滑肌为额外的开辟新的途径干预哮喘的药物治疗。
- 收到了2005年4月21日。
- 接受2005年7月22日。
- ©人期刊有限公司
引用
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}