





SARS-CoV-2造成毁灭性影响5.5亿多人感染2022年7月,大约640万人死亡(1]。社会和经济的影响将波及多年来,不断进化的SARS-CoV-2持续通过人口以减少活动传播的疫苗和单克隆抗体οBA.4或BA.5 subvariants [2]。更大的理解发病机理,因此更多的量身定制的治疗方法至关重要。
发病机理的核心已经从上呼吸道疾病的传播下呼吸道,由于缺少上呼吸道粘膜免疫控制病毒易位。在严重的情况下,这将导致低氧血症和呼吸衰竭。发病机理类似于急性呼吸窘迫综合征(ARDS),但也显示了一些差异。事后研究已经证明了ARDS的主要特性,如弥漫性肺泡损伤,血栓形成和支气管肺炎是频繁但变量结果(3]。然而,肺间质巨噬细胞的扩张COVID-19实质发生在更大程度上,相比ARDS的流感或其他原因4];并在COVID-19 microthrombus形成更大的负担相比,流感(5]。感应的强劲和持续的肺部炎症SARS-CoV-2与威胁生命的疾病有关。事实上,临床试验平台演示了改进的生存与抗炎疗法(如。糖皮质激素和白细胞介素- 6受体阻滞剂)住院患者血氧过低的(6,7]。事后研究SARS-CoV-2峰值(S)蛋白被发现在呼吸道上皮细胞,但肺部炎症没有地图,肺的病毒持续感染、暗示自治的存在炎症电路时死于呼吸衰竭(3]。
建立的程度降低呼吸道病毒复制,支持高效的病毒感染的细胞和受体和直接病毒复制的程度与自治炎症导致肺损伤是关键问题。建立功能模型病毒复制的一个主要要求补充推理基于表达式的数据和关联研究。许多研究已经确定了低级和限制表达的血管紧张素转换酶2 (ACE2),主要涉及SARS-CoV-2细胞受体结合,在肺泡空间,但报告有差异很大的确切的表达水平。齐格勒和同事就可见一斑,推断表达式从单细胞low-copy RNA序列(scRNA-seq)数据记录比如ACE2跨膜丝氨酸蛋白酶和蛋白酶2 (TMPRSS2)涉及处理病毒S蛋白与ACE2是有问题的,因为低灵敏度的检测可能会导致低估了真正的表达和蛋白水平可能不会与转录(8]。多重刺激诱导ACE2表达在人类肺癌,其中包括1型干扰素(IFN-1s) [8)或吸烟9]。此外,其他受体(如。Basignin (BSG) Asialoglycoprotein受体1 (ASGR1) Kingle-containing跨膜蛋白1 (KREMEN 1)和蛋白酶(如。组织蛋白酶L, Furin)允许SARS-CoV-2细胞输入的基础上他们的交互与相关冠状病毒在体外SARS-CoV-2或在某些情况下的研究在活的有机体内老鼠的研究COVID-19 [10,11]。
Honzke和同事使用sc或个核武器(sn) RNA-seq表明ACE2只是表示在低水平大约1.5/1000肺泡上皮类型(在)2而不是其他细胞在肺泡12]。这与更高水平的表达TMPRSS2,Furin,BSG,ASGR1,KREMEN1在一个广泛的细胞类型。重要的是,他们验证蛋白表达在组织外植体,COVID-19死后肺组织,和成人支气管瀑样干细胞。IFN-β(IFN-1亚型)未能上调ACE2在mRNA的蛋白质含量与感应。调查结果结合王和同事,发现类似的低水平的snRNA-seq ACE2的健康的肺组织,主要在at₂细胞,发现了一个低数量的染色质网站co-accessible ACE2的启动子(13]。这表明有限的网站开放的染色质,表示的一个特点独联体监管元素绑定的组合转录因子调控基因表达的时空模式限制表达式(14]。
Honzke和同事也发现SARS-CoV-2复制只有温和的肺组织的效率和效率低于甲型流感病毒(IAV)或中东呼吸系统综合症冠状病毒(MERS-CoV)。SARS-CoV-2复制只是观察到支气管瀑样(缺乏AT2-like细胞组成ACE2操纵基因诱导后表达)ACE2表达式。同样,在肺泡装有AT2-like瀑样细胞表达ACE2在低数量,低级病毒复制是局限于ACE2+细胞。一个强大的胞质染色模式特定于ACE2的病毒+细胞在两种ACE2+支气管和肺泡瀑样。相比之下,肺泡巨噬细胞(AM)病毒蛋白质或RNA只有点状的染色,这归因于作者吞噬/ efferocytosis at₂细胞或病毒的内吞作用。相反,adenoviral转染ACE2在肺移植组织扩大细胞趋向性。在事后COVID-19肺标本,证据表明病毒与病毒复制是罕见的稀有at₂细胞和低级病毒在巨噬细胞染色符合病毒吸收而不是富有成效的感染。总之,调查结果显示ACE2的受体是富有成效的感染,仅限于偶尔at₂细胞,肺巨噬细胞病毒但不支持病毒复制。然而,其他一些研究表明肺巨噬细胞可以支持病毒复制。Sefik和同事发现了病毒复制的标志(病毒RNA-dependant subgenomic RNA双链RNA, RNA聚合酶(RdRp))在人性化的小鼠肺巨噬细胞感染SARS-CoV-2,验证RdRp发现在事后人类肺部15]。格兰特和同事确认了反义病毒RNA在来自支气管肺泡灌洗(BAL)插管患者严重COVID-19 [16]。不同结果的原因尚不清楚。然而,Sefik和同事发现ACE2封锁极大地降低了病毒被巨噬细胞吸收,而Honzke发现如果肺巨噬细胞有强迫的表达ACE2他们成为有效的感染,建议没有ACE2在肺巨噬细胞表达模型研究支撑Honzke的发现。Sefik还发现了潜在的病毒通过CD16锁定吸收,有助于吸收巨噬细胞的病毒颗粒。Sefik和同事也暗示感应感染巨噬细胞(pyroptosis限制病毒复制的15]。因此,它仍然是合理的,复制的肺巨噬细胞感染是罕见的如果它发生,而巨噬细胞激活的主要驱动因素是S蛋白的检测,低fucosylation anti-S Fc地区的免疫球蛋白抗体,病毒RNA在病毒和间接摄取细胞病毒诱发的应激,感觉到通过检测DNA的cGMP-AMP synthase-stimulator干扰素基因的途径或其他炎症通路(17- - - - - -19]。
这些有趣的结果表明,肺泡病毒复制不是肺泡损伤的主要因素。Honzke和同事开采单细胞和散装转录数据从他们的感染肺外植体和事后COVID-19肺组织说明肺巨噬细胞表明病毒吸收。在体外SARS-CoV-2 MERS-CoV和冠状,从IAV诱导不同的是转录反应,突出感应IFN-stimulated基因(研究小组)和炎症反应在细胞与病毒吸收。的损失是由炎症巨噬细胞及其替代SARS-CoV-2感染外植体和事后肺,与在人性化的小鼠的发现一致(20.]。激活巨噬细胞也有集群和MERTK人口+monocyte-derived巨噬细胞。病毒内吞作用是炎症巨噬细胞,巨噬细胞的人口大多数相关研究小组的表情。也有证据表明早期诱导IL-1β和趋化因子(如。CCL3 CCL20, CXCL5 CXCL8)炎性巨噬细胞。建议激活巨噬细胞可能摄入感染at₂efferocytosis细胞,与病毒和细胞吸收与增强有关NF -κB,肿瘤坏死因子和il - 1表达式。有感应的inflammasome活化剂P2RX7 IL-1β也将帮助处理。这些数据因此强烈支持应对病毒的早期炎性巨噬细胞反应的产品。虽然在肺泡病毒复制可能不是直接受伤的司机,病毒载量与系统性炎症和临床结果,支持这种巨噬细胞反应的概念是"病毒载量(成正比21]。
这个重要的研究为未来的调查引发了许多问题。虽然功能的后果ACE2基因的方法进行了探讨,巨噬细胞的直接作用和具体的炎症通路在推动肺泡损伤需要确认操作的相关模型。在最近出版的人性化COVID-19小鼠模型,证实SARS-CoV-2诱导减肥,肺部病理学,研究小组感应与炎性巨噬细胞扩张和monocyte-derived巨噬细胞,并减少CD206+我(20.]。地塞米松和早期治疗管理SARS-CoV-2马伯减少viral-associated扰动在巨噬细胞的数量与改善的结果。的确切身份激活和炎症巨噬细胞数量大概会需要进一步描述。Rendeiro和同事发现,间质单核细胞来源的巨噬细胞的主要人口扩大COVID-19肺(4]虽然monocyte-derived巨噬细胞主导BAL严重COVID-19 [22),出现严重的IAV感染不同的特性。的相对贡献最近招募巨噬细胞来源于单核细胞或间质巨噬细胞复制的肺间质巨噬细胞扩大人口?有证据的复制和重组的居民是炎症的利基COVID-19肺部炎症和激活巨噬细胞表型应对病毒?这些问题可能会受益于lineage-tracing方法和测量原位复制。微分个体发生学或环境的变化在多大程度上改变巨噬细胞可塑性支撑COVID-19-related肺泡巨噬细胞表型(23,24]?我损失机制需要澄清。细胞凋亡通路在Honzke调节的研究,但这是损失的主要机制是吗?Sefik和同事展示了NLRP3 inflammasome肺巨噬细胞的激活和pyroptosis包含病毒,这可能是一个额外的机制的细胞损失(15]。基因变异的DPP9inflammasome,消极的监管机构,与COVID-19与重要疾病的风险25]。快速大规模损失的机制虽然居民是几天内感染(20.),一个在K18找到我们也观察到ACE2转基因小鼠(麦克休和Dockrell未发表的数据),表明失去最初的原始胎儿liver-derived人口长寿的卵黄囊。该病毒识别途径调节巨噬细胞的炎症反应吗?是什么改变了氧化应激和线粒体功能障碍的作用?最近,线粒体裂变,改变线粒体氧化磷酸化、活性氧生成和缺氧诱导factor-1α表达已确定在COVID-19肺巨噬细胞(26]。
除了这些问题之外,重要的是要考虑如何在肺泡巨噬细胞数量的变化空间,未来和持续的炎症环境,印在巨噬细胞反应。有塑性损失吗?有影响未来的感染,在巨噬细胞表型改变影响炎症和组织修复与后果的决议对肺纤维化的发展吗?IAV感染小鼠,monocyte-derived巨噬细胞的招募到肺泡空间持续一段时间后,但是他们的表型逐渐恢复到一个类似于原始居民,但很久之后解决感染(27]。是这种转变的动力学在COVID-19长时间吗?此外,Wendisch和同事确认CD163积累+与profibrotic monocyte-derived巨噬细胞转录类似profibrotic巨噬细胞的表型在特发性肺纤维化。这种表型表达的程度MERTK是因为直接吸收的病毒或改变巨噬细胞个体发生一个悬而未决的问题是最近争论(28]。无论所有的问题模拟Honzke的手稿,这项研究表明在我人口抵消扰动稳态控制施加的居民是健康的肺部炎症,促进自治电路,可以解释肺泡损伤严重COVID-19直接病毒诱导细胞病理学(独立的图1)。
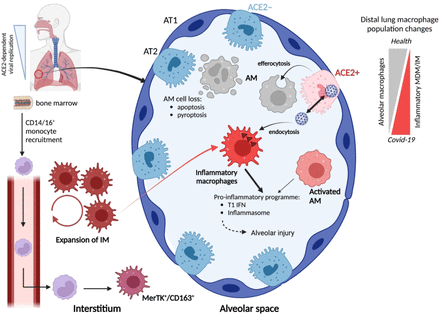
摄动远端肺巨噬细胞人口在Covid-19肺泡损伤的发病机制。AT1: 1型肺泡上皮细胞;at₂:肺泡上皮细胞2型;问:肺泡巨噬细胞;MDM: monocyte-derived巨噬细胞;即时通讯:间质巨噬细胞。
资助者:d.hD和B。米are supported by the UKRI-MRC SHIELD consortium (MR/N02995X/1) and UKKRI-MRC Programme grant (MR/W028506/1). C.D.R. is supported by an Edinburgh Clinical Academic Track (ECAT)/Wellcome Trust PhD Training Fellowship for Clinicians award (214178/Z/18/Z). UK Research and Innovation; DOI: http://dx.doi.org/10.13039/100014013; Grant: MR/N02995X/1, MR/W028506/1; Wellcome Trust; DOI: http://dx.doi.org/10.13039/100010269; Grant: 214178/Z/18/Z.
脚注
利益冲突:David H Dockrell演讲酬金和旅游欢悦的支持医疗报告;收到葛兰素史克化合物;在提交工作;和作为人类药物(MHRA)英国专员。
利益冲突:布莱恩·麦克休已收到葛兰素史克化合物以外的提交工作。
利益冲突:所有其他作者没有披露。
- 收到了2021年10月15日。
- 接受2022年8月16日。
- 版权©2022年作者
这个版本分布在创作共用署名非商业性许可证的条款4.0。商业生殖权利和权限接触权限在}{ersnet.org











{kind=link}
{kind=link}