





摘要
我们评估了间歇性缺氧(模拟阻塞性睡眠呼吸暂停(OSA)的特征之一)是否会在小鼠模型中导致粪便微生物群的改变。
在活的有机体内测定了4只麻醉小鼠间歇缺氧时结肠粪便中的氧分压。10只小鼠遭受慢性间歇性缺氧(5%氧浓度下20秒)2和40秒在室内空气6小时·日−1), 10只小鼠作为常氧对照组。通过16S rRNA焦磷酸测序和微生物生态学定量观察的生物信息学分析,获取粪便样品,确定微生物组组成。
在肠上皮近端(<200 μm)的粪便中,间歇性缺氧暴露转化为缺氧/再氧模式。间歇缺氧对全球微生物群落结构有显著影响。间歇缺氧增加了α-多样性(Shannon指数,p<0.05),引起了肠道菌群的变化(ansim分析β-多样性,p<0.05)。特别是,与对照组相比,间歇性缺氧暴露小鼠的厚壁菌门丰度更高,拟杆菌门和变形菌门丰度更低。
模拟OSA的间歇性缺氧改变了粪便微生物群的组成和多样性,表明在OSA中宿主和肠道微生物群之间的生理相互作用可能被解除。
摘要
在模拟OSA的小鼠模型中,间歇性缺氧改变了粪便微生物群的组成和多样性http://ow.ly/ERjA9
介绍
阻塞性睡眠呼吸暂停(OSA)因其高患病率已成为一个非常相关的公共卫生问题(如。30至49岁男性占10%)[1,但更重要的是,除了降低生活质量和增加交通事故,OSA还会带来重要的中长期后果,即心血管、代谢、认知和癌症相关改变。值得注意的是,考虑到超重/肥胖与OSA密切相关,预计全球患OSA的患者数量将进一步增加[2以及发达国家和发展中国家肥胖症流行的演变趋势。在OSA患者经历的生理损伤中,睡眠结构的破坏,交感神经激活增加和间歇性缺氧,后者似乎发挥了主要作用,因为它会引发炎症和氧化应激级联,这是有害的,并导致了OSA的多器官病态后果[3.].
然而,在不同的器官和组织,如大脑、肝脏、睾丸、脂肪或肌肉中,已经研究了缺氧/再氧的大小和病理生理影响[4- - - - - -6,但没有人注意到阻塞性睡眠呼吸暂停(OSA)所表现出的反复氧去饱和对肠道微生物群的潜在影响。事实上,哺乳动物的肠道是由一个由宿主生理决定的动态环境中由厚壁菌门和拟杆菌门的专性厌氧生物主导的复杂而密集的微生物群落组成的[7].此外,各种证据也表明肠道内有氧和兼性厌氧细菌的动态配置和肠道内厌氧环境的稳定性[8,9].由血氧含量的偶发性变化引起的肠道菌群改变可能与此相关,因为代谢改变,如由肠道菌群调节的肥胖和代谢综合征,通常与OSA有关[10,11].事实上,大多数OSA患者都是肥胖的,大约一半的患者同时患有代谢综合征[12].有趣的是,新的数据也支持OSA的观点本身可能反馈到导致肥胖发展或加重的机制中[13].
目的是生成初始阻塞性睡眠呼吸暂停综合症的证据揭示在这未知的方面,我们提出间歇动脉低氧血描述阻塞性睡眠呼吸暂停综合症会导致肠道微生物组内缺氧/ re-oxygenation自行车事件,因此,肠道微生物的生物多样性可能会被修改。这一假设的基本原理是基于这样的证据:肠道上皮表面的毛细血管壁,由于其主要功能是在食物消化过程中从肠道内容物中吸收营养物质,所以非常具有渗透性,显然允许氧气扩散。事实上,众所周知,虽然肠道内容物的核心是缺氧的,但在肠道上皮附近,微生物组的氧浓度有一个≈150-200µm的梯度[8].因此,最近对小鼠和人类的肠道微生物的研究报告存在与宿主组织提供的氧气和营养素分布的微生物的径向梯度存在。在这种情况下,高压氧治疗改变了小鼠中肠道微生物的组成。在人类中,与粪便相比,16S rRNA基因分析显示出与直肠粘膜相关的促菌的耐氧性生物比例增加,表明氧气浓度对微生物群的影响[9].因此,我们有理由假设,由于扩散,与OSA相关的毛细血管中氧含量的间歇性变化会引起微生物组中氧水平的波动。
为了测试我们的假设,我们对暴露于慢性间歇性缺氧模型模型的小鼠进行了研究。首先,我们测量粪便内氧气的部分压力,以确认间歇性动脉低氧血症转化为微生物组内的缺氧/再氧化事件。其次,我们对慢性间歇性缺氧和常氧对照小鼠进行了粪便微生物的分类分析,分析了肠道中的潜在微生物组改变。
材料和方法
动物
这项研究是在24只6岁的无病原体C57BL/6雄性小鼠上进行的 周(查尔斯河实验室,法国阿尔伯勒河畔圣日耳曼) 在研究开始前几天和6周的研究期间,将动物关在标准笼中,并接受自来水和消毒标准食品随意.它们被保存在动物设施的一个温度和光线都受控的房间里。实验程序由巴塞罗那大学(Barcelona, Spain)动物研究伦理委员会批准。
体内评估肠内氧气分压
第一组实验的重点是评估呼吸间歇性低氧气体混合物是否会在微生物水平上转化为缺氧/再氧引起的变化。间歇性低氧气体混合物模拟了导致阻塞性睡眠呼吸暂停(OSA)中典型氧合血红蛋白去饱和的氧气组分。因此,氧的分压(PaO2)在四组麻醉(尿烷20%,1 g·kg)的小肠粪便中测定−1),用脉搏血氧仪(MouseOx Plus;斯塔尔生命科学公司,奥克蒙特,宾夕法尼亚州,美国)。麻醉后,打开腹壁,露出肠子。在小肠壁上做一个小切口,插入快速反应克拉克极谱氧微电极移液管(OX-50;Unisense A/S,奥尔胡斯,丹麦)(直径50 μm, 90%响应时间<2秒),连接到一个放大皮安计(Unisense A/S),此前在100% O的水中校准2, 21%的人啊2记录无氧溶液(NaOH 0.1 M,抗坏血酸钠0.1 M)和氧分压(MicOX软件;Unisense / S)。微电极传感器的尖端通过微控制驱动系统通过小肠的粪便内容物逐步前进,直到到达传感器插入位置对面的肠上皮。然后,间歇性缺氧(室内空气:40秒;5%啊2空气:20秒)通过密封的鼻罩。随后,逐步取出微电极,记录离肠上皮不同距离的氧分压。
慢性间歇性缺氧的应用
对于间歇性缺氧暴露,称重10只小鼠,将其置于长26厘米、宽18厘米、高6厘米的盒子中,用空气冲洗,氧含量循环变化(40秒的室内空气和20秒的5% O的低氧空气)2),在其他地方有详细描述[6].这种间歇性缺氧的速率相当于60次呼吸暂停·h−1典型重度阻塞性睡眠呼吸暂停(OSA)患者,疗程6 h·d−1(09:00-15:00),为期6周。10只老鼠称重后被放在一个类似的盒子里,持续用室内空气冲洗(常氧对照组)。经过6周的间歇缺氧(或常氧)后,对小鼠称重,并直接从人工处理小鼠的粪便排出中获得粪便样本,并立即在−80°C下冷冻并保存至分析。在研究结束时,对动物进行麻醉(20%聚氨酯,1 g·kg)−1),然后通过腹主动脉放血安乐死。
从粪便样本中提取DNA
使用QIAamp DNA stool Mini试剂盒(Qiagen, Hilden, Germany),按照制造商的说明从200 mg粪便材料中提取DNA。DNA浓度通过260 nm (A260)的吸光度测定,纯度通过纳米滴分光光度计(Nanodrop Technologies, Wilmington, DE, USA)测定A260/A280的比值来估计。
16S rDNA序列PCR扩增及分析
16S序列是大多数微生物物种中存在的标记基因,但序列变异,允许在不同的分类学基团之间准确分离[14]使用TaKaRa Ex Taq PCR混合物(TaKaRa Bio USA,Madison,WI,USA)和PCR引物HDA1(5′-GACTCCTACGGAGGGCAGAGT-3′)和HDA2(5′-GTATTACCGCGGGCTGGCAC-3′)扩增16S rDNA基因的可变区2-3。正向引物设计为适配器A序列(CGTATCGCCTCGC GCCA)加上键序列(TCAG),反向引物设计为适配器B序列(CTATGCCCT GCCAGCCG)加上键序列(TCGA)。454个适配器(美国印第安纳波利斯罗氏应用科学公司)包含在前向引物中,然后是10个 bp样本专用多路复用标识符。PCR程序设置如下:95°C下10分钟 在95°C温度下至少进行30次循环,持续1小时 最低温度,50°C,持续1小时 最低温度和72°C,适用于1.5 最低温度,然后72°C持续10分钟 琼脂糖凝胶电泳后,使用Agencourt安瓿试剂盒(意大利米兰贝克曼库尔特)对PCR产物进行两次纯化,并使用Quant iT PicoGreen双链DNA分析试剂盒(加拿大伯灵顿Invitrogen)进行定量。根据制造商使用钛化学仪器(罗氏应用科学)的协议,在GS Junior 454平台(罗氏应用科学)中进一步处理和测序之前,获得等摩尔池。
生物信息学分析
使用定量见解进行分析454个焦点测序数据集进入微生物生态(QIIME)1.8.0软件(http://qiime.org.).原始读取根据454放大器处理管道进行初步过滤。通过QIIME的split_library.py脚本对焦序列读取进行解复用并进一步过滤。为了保证更高水平的准确性,如果读的平均质量分数小于25,并且有模糊的基调用,则从分析中排除。用于分析16S基因reads的流水线分析方法如下:对通过质量过滤器的序列进行去噪,剔除单例。通过相似性为>97%的聚类序列,筛选出操作分类单元(OTUs),并将每个聚类中最丰富的代表性序列提交uclust共识分类学作业(http://drive5.com/usearch/manual/uclust_algo.html.),利用Greengenes 16S rRNA基因数据库(http://greengenes.lbl.gov/cgi-bin/nph-index.cgi).如前所述,通过QIIME评估α-和β-多样性[15].以1%丰度过滤的OTU表通过QIIME生成OTU网络,并构建二部图,其中每个节点代表一个样本或一个细菌OTU。绘制样本和OTU之间的连接,边权定义为每个样本中出现的每个OTU的序列数。使用Unweighted UniFrac距离矩阵通过QIIME的compare_categories.py脚本进行ANOSIM统计检验。用折刀系数计算每个样品的微生物群落之间的距离。采用不加权对组算法(UPGMA)聚类树来描述QIIME生成的多个样本之间的不相似性。
统计分析
考虑时间的体重变化采用双向方差分析(前)与在6周的时间后)和治疗(间歇性缺氧与normoxia)。组间OTUs的相对丰度比较采用Wilcoxon检验,并使用软件包Explicet进行连续性校正,该软件包专门用于分析微生物组数据[16].α-和β-多样性通过QIIME实现:α-多样性使用非参数t检验,默认数量为999个蒙特卡罗排列,β-多样性使用ANOSIM统计方法,默认数量为99个排列。p值<0.05被指定为具有统计学意义。
结果
PaO2在肠腔内测量
间歇性缺氧暴露和由此导致的动脉血氧饱和度的间歇性波动,转化为靠近肠道上皮的微生物群所经历的缺氧/再氧事件。正如预期的那样,在正常的氧气呼吸中PaO2在靠近小肠中心的粪便内(即。远离上皮毛细血管)几乎为零。当血氧计探针尖端接近肠壁时,PaO2逐渐增加到〜50mmHg的值,对应于记录在肠上皮附近的那些。间歇性缺氧的应用导致了明显的波动PaO2在100-200µm范围内,随着微电极尖端向粪丸中心移动,振荡幅度减小,如图1.这个数字包括的值PaO2在应用间歇缺氧前的基线条件下(动物呼吸常氧室内空气)测量。这些数据表明,如预期的那样,在常氧条件下,粪便中的氧气梯度,而当动物遭受间歇性缺氧时,PaO2体验随着平均水平的降低而变化。
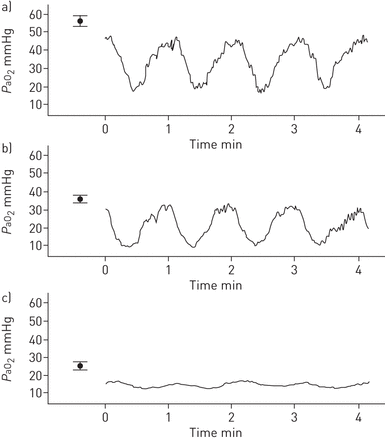
氧分压的例子(PaO2)在小肠内。时间的PaO2小肠记录在管腔内的三个点进行间歇性低氧模拟阻塞性睡眠呼吸暂停时。a)紧邻肠上皮;b)向肠腔中央100 μm; c)向肠腔中央200 μm。对于每个点,符号表示平均值±sd间歇缺氧前测量的2分钟基线(常氧)值。
变化体重
均值±sd正常缺氧组和间歇缺氧组的基线体重分别为22.11±0.52 g和22.71±0.30 g。6周后,体重分别为24.44±0.67 g和23.41±0.29 g。时间是体重增加的一个显著因素(p<0.001),而治疗(间歇性缺氧)与常氧)的差异无统计学意义(p=0.747)。
间断性缺氧和常氧相关肠道菌群多样性的估计
通过QIIME筛选的16S rRNA基因序列共88 884条,平均每个样本7407条。QIIME后所有小鼠的粪便菌群由883个OTUs组成,至少4个样品中相对丰度> . 1%。在评估α和β多样性测度之前,样本被精简为2500个序列,这与从数据集中任何单个样本中获得的最低数量的质量reads相对应。因此,每个研究组有两个样本由于质量水平较低而被排除在分析之外。间歇性缺氧组的OTUs数量高于正常缺氧组(平均284与221年,分别;p = 0.019)。各组粪便样本的平均多样性(Shannon)和群落丰富度(Chao)表明,与对照组相比,间歇缺氧暴露小鼠粪便样本的细菌多样性和丰富度有更高的趋势(表1).以3%距离计算的观察到的OTUs的稀疏曲线在~ 300 reads时开始趋于平稳,这表明较高的每个样本读取数并不会提供更全面的细菌类群目录。间歇缺氧组和常氧组的Shannon指数差异显著(p=0.010),表明物种分布的均匀性增加。换句话说,少数优势物种的丰度下降,有利于群落中的少数成员。然而,细菌丰富度的Chao1指数并没有随着reads的增加而趋于平稳,这表明在我们的深度水平下,可能无法检测到群落多样性的主要成分。基于每个样本OTUs相对丰度的unweighted Unifrac主成分分析提供了关于两个研究组粪便细菌微生物群之间系统发育关系的有用信息。在坐标组合中,常压组的所有样本都与IH组分离,两个主成分得分分别占总方差的59% (PC1)和17% (PC2)。ansim与排列确认显著的组分离(p=0.01) (图2),表明间歇性缺氧和常氧基团的微生物组成存在明显差异。Upgma树代表了间歇性缺氧和常氧小鼠的微生物群落成员的相似性。值得注意的是,间歇性缺氧和常氧小鼠彼此分开聚集,并且在间歇性和常氧群中的样品中存在明显的区别(高(75-100%)千克支援)(图3).
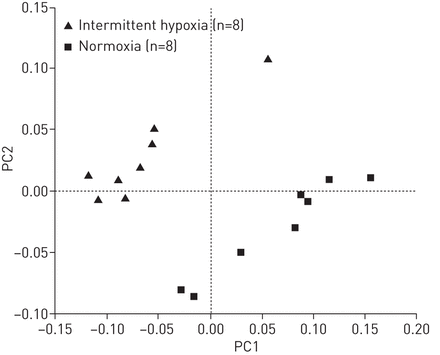
主成分分析,PC1与PC2,来自454测序的粪便样本的细菌群落。每个物种的细菌群落沿两个主坐标轴的位置,以及每个坐标轴解释的变异百分比。结果基于unweighted Unifrac指标。PC1 = 59.11%;PC2 = 17.39%。
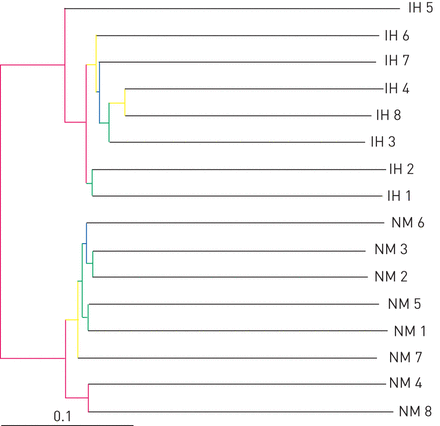
使用算术平均算法的未加权对组法使用未加权Unifrac距离矩阵进行分层聚类。颜色表示不同的jackknife支持度。红色:75–100%;黄色:50–75%;绿色:25–50%;蓝色:<25%支持度。条形表示社区差异性。IH:间歇性缺氧;NM:常氧。
小鼠粪便中间歇性缺氧和常氧16S rRNA基因焦磷酸测序
在本研究中,不同细菌水平下,间歇缺氧组和常缺氧组的粪便菌群组成存在显著差异。门水平上,大多数OTUs属于拟杆菌门(69.79%为间歇缺氧)与normoxia 73.61%;p = 0.10)。其次是厚壁菌门(22.74%的间歇缺氧)与27.63%的常氧;p = 0.49),然后是蛋白菌(1.20%间歇性缺氧与1.50%常氧;p=0.71)。剩余的细菌群属于其他七个门,在至少四个样本中相对丰度<1%(图4).在两组研究中,没有一个门表现出统计学上的显著差异。此外,对照组和间歇缺氧组间的厚壁菌门/拟杆菌门比值无显著差异(p=0.10)。

间歇缺氧组和常氧组小鼠门的焦磷酸测序分析。
各组小鼠共检出27个家族,以S24-7家族(47.70%为间歇性缺氧)为主与normoxia 53.21%;p=0.12),其次为碱性蒿科(33.33%为间歇缺氧与normoxia 43.14%;p=0.27), Desulfovibrionaceae(25.31%间歇性缺氧)与normoxia 5.01%;p=0.27)和瘤胃球菌科(20.74%间歇缺氧)与normoxia 30.29%;p = 0.37)。然而,我们发现其他9个少数民族家庭之间存在显著差异。普氏菌科(Prevotellaceae)的相对丰度(11.82%为间歇缺氧与normoxia 5.94%;p=0.01), Paraprevotellaceae(7.64%间歇性缺氧与normoxia 3.78%;p=0.03)和Lachnospiraceae(6.03%间歇缺氧)与normoxia 1.77%;P <0.001),间歇缺氧组明显高于正常缺氧组。间断性缺氧组丹毒藓科(4.23%)含量显著降低与normoxia 18.19%;p<0.001)、拟杆菌科(2.10%间歇缺氧与8.31%常氧;p<0.01),土杆菌科(0.33%间歇性缺氧)与normoxia 7.26%;p<0.001),臭杆菌科(1.34%间歇缺氧与normoxia 2.34%;p=0.04)和Peptococcaceae(0.26%间歇性缺氧与normoxia 1.96%;P =0.01)。此外,在其他丰富的科,包括卟啉单胞菌科(1.66%间歇缺氧),两组间无显著差异与normoxia 2.01%;p=0.27), Rikenellaceae(6.29%间歇性缺氧)与normoxia 7.95%;p=0.08),乳酸杆菌科(2.39%间歇缺氧与normoxia 2.45%;p=0.20)和Enterobacteriaceae(间歇缺氧1.68)与normoxia 1.67%;p = 0.65) (图5).
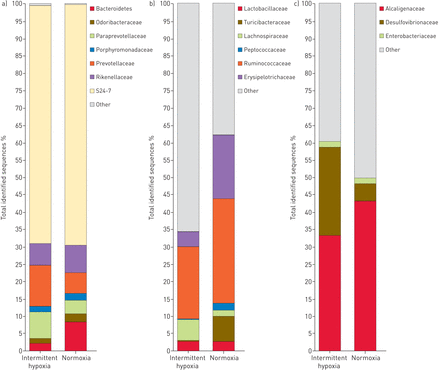
间歇缺氧和常氧粪便样本中细菌的科级微生物分类。a)拟杆菌门科;b)厚壁菌门科;c)变形菌门科。
我们还发现研究组之间在属水平上的微生物组成存在显著差异。在20只取样小鼠中,共鉴定出23个属,其中6个属在间歇性缺氧和常氧组之间存在显著差异。嗅杆菌(1.34%间歇性缺氧与normoxia 2.34%;p=0.04)、拟杆菌(2.10%间歇缺氧与normoxia 8.31%;p<0.001)、Turicibacter(1.33%间歇缺氧与normoxia 7.26%;p<0.001)和前脑(4.03%间歇缺氧)与normoxia 19.97%;P <0.001),占粪便样本细菌总数的1%以上,且常氧组细菌显著增多。此外,Paraprevotella(7.64%间歇缺氧与3.69%的常氧;P = 0.03)和PREVOTALLLA(11.82%间歇性缺氧与normoxia 5.94%;P =0.01),间歇性缺氧组的丰度明显高于正常缺氧组。另一方面,脱硫弧菌(24.89%间歇缺氧)与间歇缺氧组以常氧菌属(4.34%)最多,间歇缺氧组以苏苔菌属(33.33%)最多与43.14%常氧)和示波螺旋藻(12.76%间歇性缺氧)与常氧组以属为主。间歇性缺氧组和常氧组前三属丰度差异无统计学意义(p>0.05) (图6).
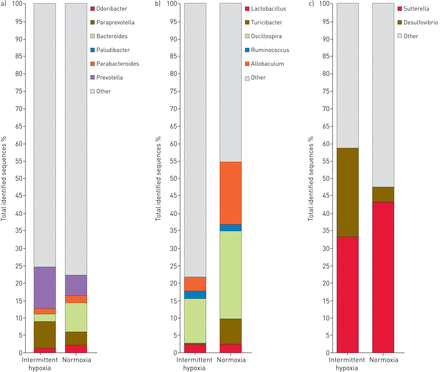
间歇缺氧和常氧暴露小鼠微生物区系中细菌属的相对丰度。a)拟杆菌门属;b)厚壁菌门属;c)变形菌属。
讨论
据我们所知,这是第一个研究间歇性缺氧模拟OSA对肠道微生物组成的影响。在这里,我们结合了肠道(小肠粪便)内氧浓度的测量和粪便微生物组的群落水平分类分析,以确定氧张力的电位振荡如何影响宿主的肠道微生物群。通过对间歇缺氧和常氧小鼠粪便16S rDNA基因标记的高温测序分析,发现在门、科和属水平上优势类群的相对丰度存在较大差异。
对变化的评估PaO2由肠内的间歇性缺氧诱导的,用氧传感器进行,这是以前用来测量的PaO2阻塞性睡眠呼吸暂停动物模型的不同组织[4,6].正如预期的那样,当传感器接近肠道内容物的缺氧核心时,粪便中的氧合水平下降。然而,值得注意的是PaO2可在以下范围内检测到振荡:∼0–200 肠上皮表面的µm,表明微生物群的相关部分易受间歇性缺氧引起的氧合变化的影响。这些实验数据与半无限介质中Fick第二扩散定律解的理论预测一致(模拟上皮表面粪便内的自由扩散)。事实上,距离表面z处氧浓度振荡的振幅(A)为:A(z)=A0·e−π·f·z/D式中f为振荡频率,D为氧在介质中的扩散系数。考虑到粪便中的D是2×10−5 厘米2·年代−1,类似于水和其他生物组织的值[17],呼吸暂停发生频率为60次·h−1,得到A(z)/A0 =e−z / (195µm)这表明在离上皮表面100、200和300µm处的振幅PaO2振荡分别从100%减少到60%、36%和21%。虽然这一理论预测没有考虑需氧微生物的潜在耗氧量,但它使我们能够解释测量数据,清楚地表明,模拟OSA的间歇性缺氧实际上转化为肠道内的环境变化,并可能被肠道微生物群感知。
与之前对肥胖、1型和2型糖尿病或克罗恩病的观察结果相反[18- - - - - -21],我们在间歇缺氧组和常氧组比较时,未发现肠道菌群在门水平上有显著差异。相比之下,我们发现在间歇性缺氧组的粪便中优势菌科和属水平上存在显著差异:普雷沃氏菌(Prevotella)、副aprevotella、Desulfovibrio、Lachnospiraceae增加,而Bacteroides、odor bacter、Turicibacter、Peptococcaceae、erysipelotricaceae减少。这些发现表明,在间歇性缺氧暴露后,优势菌群的科和属有所不同,在间歇性缺氧组中,专性厌氧菌相对富集。与正常缺氧相比,动物在间歇性缺氧时肠道内含氧量的总体减少(图1)将赋予厌氧菌一种生态选择优势,使其更具竞争力,并使其能够过度生长。相反,兼性厌氧菌甚至需氧菌将处于不利地位。值得一提的是,所分析的粪便样本不仅来自肠上皮附近缺氧/再氧化最明显的区域,还来自肠腔的内部区域,该区域具有更连续的非常严重的缺氧模式。
最近,已经确定了三个被称为“肠道型”的强大集群,它们不是国家或大陆特有的。将一个微生物组划分到给定的肠型是根据其相对富集到三个属之一:拟杆菌属(肠型1)、普氏菌属(肠型2)或瘤胃球菌属(肠型3)[22].常氧和间歇性缺氧基团可以被分类为晶体型1和2,鉴于其相对富集在Bacteroides和Fvototella,以及间歇性缺氧微生物群中脱硫弧菌丰度的显著增加[22].在间歇性缺氧组中发现普雷沃氏菌和脱硫弧菌的同时存在,表明粘蛋白降解生态位的进化,在普雷沃氏菌介导的粘蛋白降解过程中释放的硫酸盐被脱硫弧菌清除,这一过程需要防止硫酸盐抑制粘蛋白降解[22,23].粘蛋白被细菌降解通常被认为是发病的初始阶段,因为它会破坏宿主粘膜表面的保护。在我们的研究中,在间歇性缺氧组中普雷沃氏菌(Prevotella)和脱硫弧菌(Desulfovibrio,黏液降解微生物)丰度的显著增加可能表明肠道上皮层缺乏这种黏液,这可能导致肠道通透性的显著改变。此外,富集的Desulfovibrio细菌通过减少硫酸盐产生硫化氢,而硫化氢被报道为可能导致结直肠癌的危险因素[24,25].
专性厌氧Lachnospiraceae家族的一些成员在间歇性缺氧小鼠中显著富集,此前已发现与肥胖有关[26和产生丁酸的能力[27,一种对微生物和宿主上皮细胞生长都很重要的物质。此外,我们的结果还表明,与正常缺氧对照相比,间歇性缺氧小鼠的细菌多样性和聚集度分别增加,这在主坐标图和UPGMA树形图中得到了明确的显示。Shannon多样性指数表明,间歇性缺氧时肠道细菌多样性的增加,可能会改善微生物的完整性,并增加保护人类肠道免受环境胁迫的能力,如肠道内氧水平的变化。尽管肠上皮细胞对缺氧具有显著的抵抗能力,但吸收和屏障上皮细胞的功能是受氧调节的[28].事实上,缺氧/复氧会直接损害细胞功能通过渗透率和细菌移位增加,紧密连接完整性降低[29].最近,Kheirandish- g奥扎尔等.[30.]的研究表明,与健康对照组相比,患有阻塞性睡眠呼吸暂停综合症的儿童,即使是那些不肥胖的儿童,也表现出明显较高的血浆脂多糖结合蛋白水平。这些结果支持这样一种可能性,即肠道菌群的改变可能会促进肠道通透性的改变,或者,诱导微生物易位,所有这些都可能导致全身脂多糖水平的增加。循环内毒素水平的增加将促进先天免疫事件,触发低级别全身炎症过程,这可能导致代谢功能障碍。
总之,尽管不能排除在间歇性缺氧小鼠中观察到的微生物群的改变可能是部分,部分地,对于能够改性微生物群的宿主中间歇性缺氧引发的全身免疫途径,但它是合理的假设实际报告的变化PaO2暴露于真实模拟间歇性低氧血症(OSA特征)的小鼠粪便微生物群中,肠道内驱动了专性厌氧菌丰度的变化和多样性的增加。证实了间歇性缺氧对小鼠整体微生物群落结构有显著影响的假设,提示OSA患者宿主与肠道微生物群落之间的稳态关系可能受到破坏。本研究在睡眠呼吸暂停领域开辟了一个新的研究领域,对于间歇性缺氧和睡眠碎片化的影响无疑值得进一步研究[31[肠道微生物肿瘤及其在动物模型和患者中对宿主代谢的调节。
脚注
支持声明:由西班牙经济和竞争力和竞争力(SAF2011-22576)有支持这项工作。研究小组属于Instituto de Salud Carlos III(西班牙马德里)的Centros deInvestigaciónNen红色(Ciber,CB06 / 03/0018)。I. Moreno Indias由Sara Borrell博士后合同(CD12 / 00530)提供支持。M.I.Queipo-Ortuño承认Miguel Servet I型计划的支持(CP13 / 00065)和F.Cardona承认从Miguel伺服型II计划(CP13 / 00023)的支持,从Instituto de Salud Carlos III中得到支持。
利益冲突:无声明。
- 已收到2014年10月6日。
- 接受2014年11月19日。
- 版权所有©2015











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}