





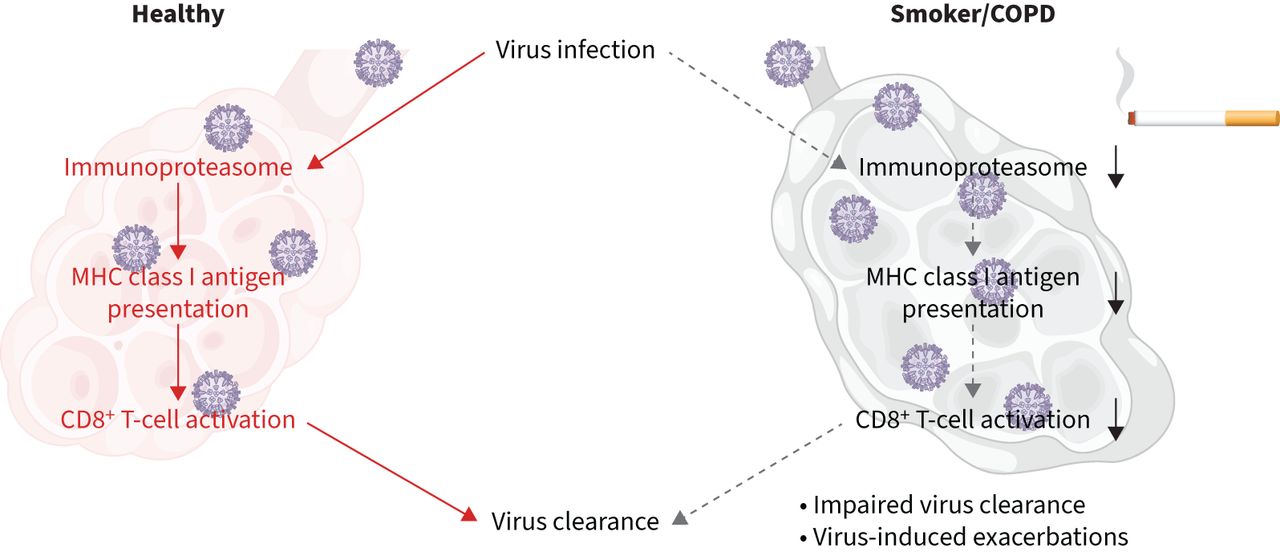
图形抽象
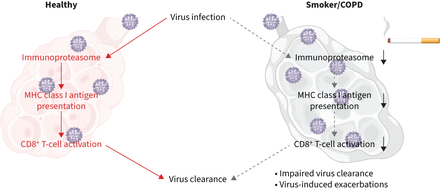
这项研究的主要发现。吸烟损害upregulation占据的主要组织相容性复合体(MHC)类我抗原演示机器导致抗病毒CD8的激活+t细胞。
文摘
背景病毒感染慢性阻塞性肺病急性加重和恶化。抗病毒免疫激活病毒特异性CD8中心+t细胞的病毒抗原表位在主要组织相容性复合体(MHC)类分子受感染的细胞。这些抗原表位immunoproteasome生成的,专门的细胞内蛋白质降解机,在感染细胞抗病毒细胞因子诱导的。
方法我们分析了香烟烟雾对细胞因子的影响,virus-mediated immunoproteasome的感应在体外,体外和在活的有机体内利用RNA和蛋白质印迹分析。CD8+t细胞激活决心在培养化验香烟smoke-exposed甲型流感病毒(IAV)来华的细胞。Mass-spectrometry-based我一定肽MHC类分析揭示了香烟烟雾对炎症的影响在肺细胞抗原表达。IAV-specific CD8+患者外周血中t细胞数量确定使用四聚物的技术。
结果吸烟损害immunoproteasome通过细胞因子信号的感应和肺细胞的病毒感染在体外,体外和在活的有机体内。此外,香烟烟雾改变了肽的抗原在MHC类分子在炎症条件下。重要的是,MHC类I-mediated IAV-specific CD8的激活+t细胞抑制了香烟。慢性阻塞性肺病患者表现出循环IAV-specific CD8数目减少+t细胞与健康对照组和哮喘患者。
结论我们的数据表明,香烟烟雾干扰我类MHC抗原生成和演示,从而有助于激活CD8受损+在病毒感染t细胞。这增加了重要的机械的见解如何吸烟介导的易感性增加吸烟者和慢性阻塞性肺病患者病毒感染。
Tweetable文摘
吸烟损害upregulation占据我抗原MHC类的演示机器导致抗病毒CD8的激活+t细胞。这可能减少病毒间隙和增加对病毒在慢性阻塞性肺病急性加重。https://bit.ly/43o0p3D
介绍
慢性阻塞性肺病的特点是进步的气流限制和肺泡的破坏,导致肺功能下降和严重降低生活质量,1]。>世界上10%的人口被诊断为慢性阻塞性肺病的死亡使得全球慢性阻塞性肺病死亡的第三大原因(2]。烟草消费是发展中慢性阻塞性肺病的主要危险因素之一(1]。突然发作的疾病,肺功能下降相关病毒感染,如鼻病毒和甲型流感病毒(IAV)或细菌3,4),导致慢性阻塞性肺病恶化[5,6]。
在抗病毒免疫中起到一个关键球员是细胞毒性CD8的激活+t细胞专门杀死感染病毒的细胞。快速和有效的CD8的重要性+t细胞激活的病毒感染的控制和解决目前再发现的冠状病毒疾病2019例(7,8]。严重急性呼吸系统综合症冠状病毒2 (SARS-CoV-2)有效地削弱被感染的细胞产生干扰素的能力,在生理条件下局部限制病毒的复制和立即报警的先天免疫系统9]。不受阻碍的病毒复制驱动的先天免疫反应过度膨胀导致hyperinflammatory肺部免疫病理反应而保护t细胞反应延迟(10,11]。同样,病毒感染和慢性阻塞性肺病患者急性加重的特点是受损的病毒清除,hyperinflammatory先天免疫反应和无效的t细胞免疫(3,12- - - - - -14]。在实验小鼠模型、香烟烟雾暴露限制不仅抗病毒先天免疫反应,而且激活适应性CD8受损+t细胞(15,16),支持的认为吸烟是一个主要的驱动力受损抗病毒免疫反应在慢性阻塞性肺病17,18]。
激活的抗病毒细胞毒性CD8+实现t细胞在识别virus-derived抗原肽与主要组织相容性复合体(MHC)类我感染病毒的细胞表面分子19]。这些病毒抗原表位主要产生在细胞内病毒蛋白质的降解immunoproteasome [20.]。immunoproteasome是一种特殊类型的蛋白酶体(细胞的主要protein-degrading机械),其中包含一组诱导催化亚基,即低分子量蛋白质(LMP) 2、multicatalytic肽链内切酶复合体亚基(MECL) 1和LMP7。这些单元转录诱导病毒感染,I和II型干扰素(IFN)信号,为抗原MHC的生成类我感染病毒的细胞抗原表位20.- - - - - -22]。实验感染小鼠肺病毒引起明显诱导的三个蛋白水解亚基在肺immunoproteasome抗病毒免疫防御的内在组成部分(21,23,24]。基因敲除小鼠缺乏一个或多个蛋白水解子单元的immunoproteasome严重阻碍有效地打击病毒感染,展示的基本功能immunoproteasome抗病毒t细胞免疫(21,25]。我们以前所示抑制免疫细胞的持续表达immunoproteasome吸烟和慢性阻塞性肺病导致受损I-mediated CD8 MHC类+t细胞激活在体外和在活的有机体内(26]。在目前的研究中,我们调查是否香烟烟雾干扰细胞因子和virus-mediated感应immunoproteasome和MHC类的我肺实质细胞抗原表达及其后果。
方法
材料和方法的细节,读者被称为补充材料。
人类的样本
EDTA-blood人类白细胞抗原(HLA) a2阳性样品稳定的慢性阻塞性肺病(n = 11)和哮喘(n = 12)患者经CPC-M bioArchive综合肺病学中心(中国共产党;德国慕尼黑)。肺健康献血者的血液样本(n = 10)从亥姆霍兹慕尼黑中心被分配。所有慢性阻塞性肺病患者每年收到IAV接种疫苗。这项研究是由当地伦理委员会批准的路德维希-马克西米利安-(德国慕尼黑;伦理投票# 382 - 10)。EDTA-blood HLA-A2-positive健康吸烟者(过度吸烟者和吸烟者,n = 7)和不吸烟者(n = 7,包括一个水烟吸烟者)获得BioMaterialBank北部和当地大学的伦理委员会批准的吕贝克(德国吕贝克;伦理票14 - 225和22 - 583)。写所有研究对象的知情同意了。在研究参与者给出细节表1和补充表S1和S2。
统计分析
数据显示为图中所描述的传说。详细分析了两组对比图中传说,与统计学意义表示p < 0.05, p < 0.01, p < 0.001, p < 0.0001。统计分析了使用软件GraphPad棱镜(版本9.00)。统计评估immunopeptidome分析使用studio 2021.09.1和R版本4.1.2 (2021-11-01)。非参数双向对齐秩变换进行方差分析使用R包ARTool (0.11.1)。
结果
吸烟会损害细胞因子和immunoproteasome upregulation占据在体外,体外和在活的有机体内
我们首先验证virus-mediated immunoproteasome诱导的肺上皮细胞使用公开数据集IAV-infected老鼠。单细胞RNA-sequencing (seq)数据分析解决最突出的三个immunoproteasome感应子单元Psmb8(编码LMP7),Psmb9(编码LMP2)和Psmb10(编码MECL-1)感染肺泡上皮细胞(aec) (补充图S1a)[27]。散装RNA-seq IAV-infected老鼠原子能委员会的数据显示最高upregulation immunoproteasome子单元3天后IAV感染(补充图印地)[28]。
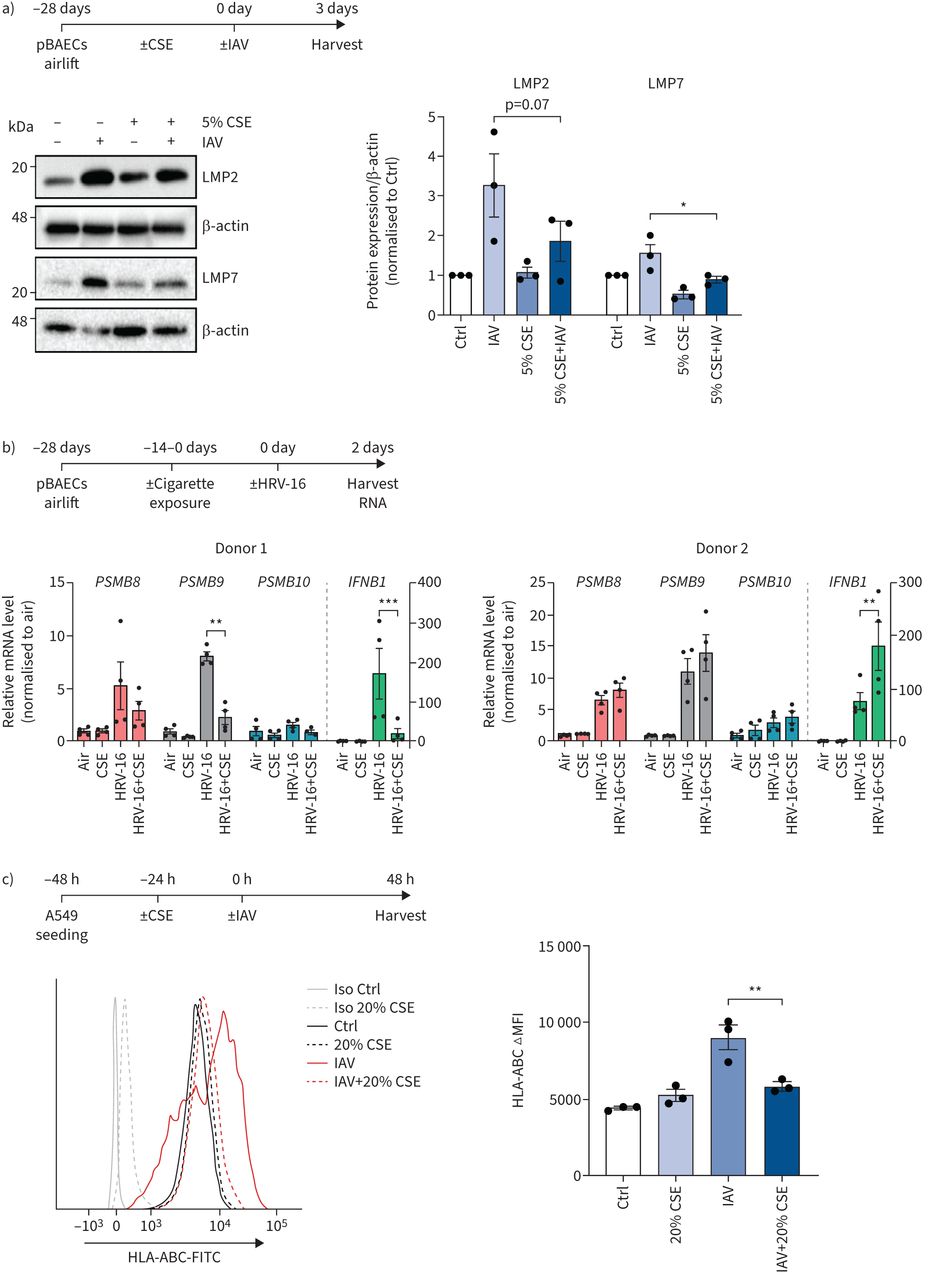
然后我们调查了香烟烟雾暴露的影响在原子能委员会immunoproteasome感应在体外。在第一组实验中,小鼠MLE-12原子能委员会是刺激与IFN-γco-treated无毒剂量的香烟烟雾提取物(CSE) (补充图1 c)[29日]。值得注意的是,25%的CSE显著抑制诱导的两个主要催化亚基immunoproteasome LMP2和LMP7后24小时(图1一个)。这种抑制作用是明显还在较低剂量的10%为所有三个immunosubunits CSE,包括MECL-1 mRNA水平(补充图S1d)。接下来,我们预处理MLE-12 CSE和转染细胞合成双链RNA (ds)模拟PolyI: C作为dsRNA病毒感染的代理。CSE剂量依赖性抑制immunoproteasome感应在co-treatment PolyI: C(1或5μg·毫升−1)在MLE-12细胞24 h后通过免疫印迹分析(如图所示图1 b)。MLE-12细胞感染小鼠γ-herpesvirus (MHV) -68年,预处理与10%或25% CSE也有效地中和病毒诱导后的immunoproteasome upregulation 72 h (图1 c)。类似的,尽管不那么明显,抑制作用的观察CSE PolyI: C -或immunoproteasome IFN-γ-mediated感应的体外肺组织治疗24小时后使用鼠标时展示肺片(图1 d,补充图S1e)。使用一个在活的有机体内香烟烟雾暴露模型与PolyI: C语言的挑战,我们解剖转录immunoproteasome激活动力学2,8,单剂后24小时10μg PolyI: C治疗。香烟烟雾暴露小鼠24天减毒的敏锐的感应Psmb8,Psmb9和Psmb108 h后PolyI: C滴剂(补充图S2a)。经过24小时的PolyI: C语言刺激,immunoproteasome亚基的蛋白质含量LMP7也显著降低小鼠肺(图2一个)。染色的肺组织部分immunoproteasome透露,PolyI: C诱导LMP2主要在内皮毛细管和支气管上皮细胞,以应对PolyI: C (补充图2 b)。第二个小鼠模型证实这些数据激活immunoproteasome占据。老鼠被暴露于香烟烟雾28天,然后感染mhv - 68 7天,蛋白表达的LMP2肺明显减毒smoke-exposed小鼠相比air-treated控件(图2 b)。随着体重,病毒负荷和lung-resident CD3的数量+和CD8+t细胞在香烟烟雾暴露小鼠相比没有显著不同控制老鼠(补充图S2c-f),减毒诱导LMP2可能不是由于感染的病毒载量或响应的改变在香烟烟雾暴露。在一起,我们的数据表明,香烟烟雾抑制PolyI: C -和immunoproteasome upregulation占据的老鼠肺部。
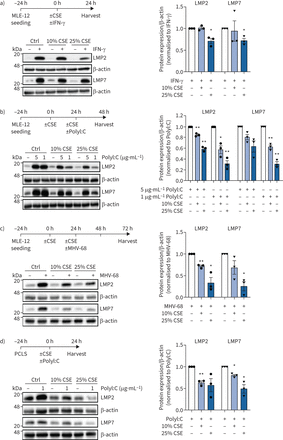
吸烟会损害干扰素(IFN) -γ和immunoproteasome upregulation占据在体外和体外。a) Immunoproteasome亚基低分子量蛋白质(LMP) 2和LMP7表达MLE-12处理10%或25%香烟烟雾提取物(CSE)和75 IU·毫升−1IFN-γ24 h。光密度分析LMP2和LMP7表达式正常化β-actin IFN-γ-treated细胞控制设置为1。b) LMP2和LMP7表达式MLE-12处理10%或25% CSE 24 h,然后用1或5μg electroporated·毫升−1PolyI: C 24 h。光密度分析LMP2和LMP7表达式正常化β-actin PolyI: C组设置为1。c) LMP2和LMP7表达式MLE-12处理10%或25% CSE 24 h,然后感染小鼠γ-herpesvirus (MHV) -68(感染复数(MOI) 1) 48 h。光密度分析LMP2和LMP7表达式正常化β-actin mhv - 68感染组设置为1。d) LMP2和LMP7表达在鼠标展示肺片(pcl)接受10%或25% CSE和培养1μg·毫升−1PolyI: C 24 h。光密度分析LMP2和LMP7表达式正常化β-actin PolyI: C组设置为1。数据意味着±扫描电镜单样本t检验;*:p < 0.05,* *:p < 0.01。
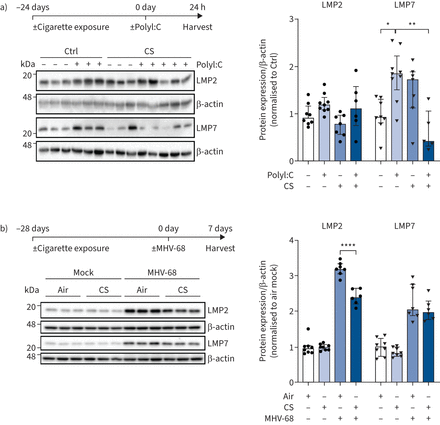
吸烟会损害PolyI: C和immunoproteasome upregulation占据体内。)低分子量蛋白质(LMP) 2和LMP7表达小鼠肺治疗后24 h 10μg PolyI: C后24天的香烟(CS)曝光。光密度分析LMP2和LMP7表达式正常化β-actin加载与控制(Ctrl)意味着没有PolyI: C治疗设置为1(中位数、四分位范围)。b) LMP2和LMP7表达在小鼠肺后28天香烟暴露和随后的感染小鼠γ-herpesvirus (MHV) -68 7天。光密度分析LMP2和LMP7表达式正常化β-actin与空气模拟控制设置为1(中位数、四分位范围)。克鲁斯卡尔-沃利斯邓恩的测试后测试;*:p < 0.05,* *:p < 0.01,* * * *:p < 0.0001。
吸烟会损害virus-mediated immunoproteasome和类MHC的感应人类上皮细胞
我们进一步分析了香烟烟雾对virus-mediated感应immunoproteasome的人类细胞。为此,我们培养的主要人类支气管上皮细胞(pBAECs)气液界面和与临床相关的病毒株感染,即。IAV或人类鼻病毒(HRV) -16。在第一组实验中,从三个不同的捐赠者pBAECs长期暴露于低剂量的香烟烟雾提取物(CSE);5%)基底外侧介质在整个过程中分化为28天(30.]。感染IAV后,我们观察到的最高感应immunoproteasome后3天(补充图S3a)。值得注意的是,CSE明显受损的IAV-mediated感应immunoproteasome子单元LMP2和LMP7 IAV (图3一)。在第二组实验中,我们从两个不同的暴露空运pBAECs捐助者(补充表S2)每天两支14天、28天的评分之间的区别(31日]。细胞随后被感染HRV-16 immunoproteasomal基因表达进行了分析使用逆转录酶病毒感染后48 h定量PCR。香烟减毒HRV-16-mediated感应的LMP2,但不LMP7和MECL-1捐赠1而不是捐助2 (图3 b)。供体特异性反应伴随着微分调节IFN-β:捐赠1对香烟烟雾减少IFN-β感应,而捐助2是由香烟烟雾暴露的影响(图3 b)。我们的数据从而证明吸烟会削弱感应的immunoproteasome pBAECs应对病毒感染可影响病毒特异性抗原MHC类我表示和CD8+t细胞激活。我们进一步分析了人类alveolar-derived癌细胞系A549和测试是否吸烟损害virus-driven感应类MHC分子。IAV-infection不仅强烈诱导immunoproteasome A549细胞,而且还调节表面表达MHC类我近两倍,由流式细胞术(图3 c和补充图S3b)。无毒剂量的CSE中和病毒诱导upregulation MHC类的我和immunoproteasome (图3 c和补充图S3b和c)。
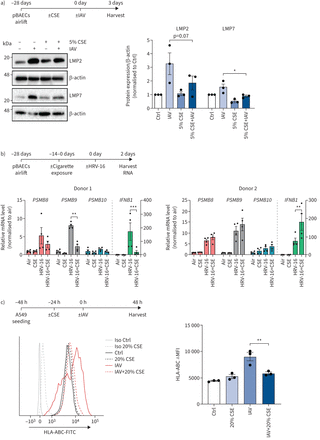
吸烟会损害virus-mediated immunoproteasome感应和主要组织相容性复合体(MHC)类我人类上皮细胞。)低分子量蛋白质(LMP) 2和LMP7表达主要人类支气管上皮细胞(pBAECs)从三个不同的捐赠者,在气液界面培养条件有或没有香烟烟雾提取物(CSE)介质(30.)和感染甲型流感病毒(IAV) 3天(60]。光密度分析LMP2和LMP7表达式正常化β-actin与治疗组设置为1。b)从两个男性健康的捐赠者pBAECs感染人类鼻病毒(HRV) -16曝光后2天至14天每天两支在气液界面分化阶段。信使rna水平PSMB8(LMP7),PSMB9(LMP2)和PSMB10(multicatalytic肽链内切酶复合体亚基(MECL) 1)IFNB1(interferon-β)测定使用逆转录酶定量PCR和相关的管家基因表达(产生HPRT和RPL32)。数据显示为四个复制每捐赠(混合两种技术和两个独立的文化),意味着±扫描电镜。c)人类白细胞抗原(HLA) abc A549细胞表面表达接受20% CSE 24 h和感染IAV(感染复数1)24 h。荧光强度显示为±扫描电镜三个独立的实验。控制:控制;Iso:同形像;FITC:异硫氰酸荧光素;ΔMFI:平均荧光强度的变化。单向方差分析Bonferroni测试后。* *:*:p < 0.05, p < 0.01, * * *: p < 0.001。
吸烟会损害IAV-specific CD8的激活+t细胞
接下来,我们分析是否吸烟也抑制了virus-mediated CD8的激活+t细胞。我们使用一个IAV-specific CD8+CD8 t细胞活化分析涉及+t细胞克隆承认IAV M158 - 66基质蛋白抗原决定基的hla a2 (图4一)[32]。同源IAV抗原决定基是由immunoproteasome生成。因此,激活的t细胞克隆效率取决于活动的immunoproteasome IAV感染(33]。主要人类肺成纤维细胞(phLF)与hla a2积极捐赠者受到CSE或空气,然后感染IAV培养IAV-specific t细胞克隆。的分泌白介素2 (IL)量化确定抗原t细胞激活(图4一)[32]。我们证实,应用剂量的CSE是无毒phLFs使用MTT检测(补充图S4a)。IAV感染强烈诱导immunoproteasome和激活CD8的表达+t细胞的分泌(- 2 5倍增加图4 b和补充图S4b)。特异性的t细胞克隆对M1肽识别证实了外部加载的phLFs M158 - 66肽(GILGFVFTL) (补充图自己)。CSE暴露显著减毒的能力IAV-infected phLFs激活IAV-specific t细胞克隆,但并未改变non-IAV-infected t细胞反应细胞(图4 b)。这些概念验证数据从而表明香烟介导的抑制immunoproteasome我类MHC抗原表达可能导致激活病毒特异性CD8受损+t细胞对病毒感染的肺细胞。
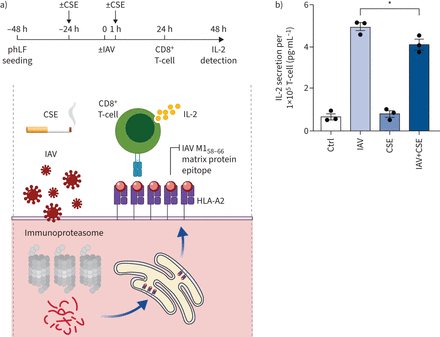
吸烟会损害激活甲型流感病毒(IAV特殊CD8)+t细胞。)示意图描述IAV的抗原表达分析:immunoproteasome-dependent处理IAV M1基质蛋白生成M158 - 66肽,装上人类白细胞抗原(HLA) a2主要组织相容性复合体(MHC)我在内质网分子。细胞表面hla a2 / M1-peptide复合物被运送到了他们激活特定的t细胞杂种瘤细胞系4 va1分泌白介素2 (IL),这可以通过ELISA检测。b) IAV-specific CD8+激活t细胞在培养与IAV-infected hla a2+主要人类肺成纤维细胞。主要人类肺成纤维细胞(phLFs)对待香烟烟雾提取物(CSE) 10% 1%胎牛血清24 h,感染IAV(感染复数1)1 h和进一步培养24小时有或没有CSE然后CD8培养+ifluenza-M1蛋白特异性t细胞的比例1:2 24 h。- 2分泌的mouse-derived t细胞克隆是从上层清液(均值±量化扫描电镜)。控制:控制。分析了意义与Bonferroni测试后进行单向方差分析。*:p < 0.05。
香烟烟雾中深刻地改变了我炎症类MHC抗原曲目
这些结果受损的MHC类我限制t细胞激活促使我们探讨香烟烟雾的影响基于pan-MHC类我抗原曲目使用质谱仪immunopeptidomics [34]。这种peptidomics方法包括下拉MHC分子,随后的MHC我一定肽洗脱和基于质谱肽识别。满足大细胞数量的需求,我们采用人类肺上皮细胞系A549和调查CSE IFN-γ-induced MHC类我限制的影响抗原。预处理的20% CSE A549细胞48 h,然后co-treated中包含的另一个24小时CSE有/没有IFN-γ。三个独立的实验进行复制与技术复制,即。质谱分析。虽然选择剂量的CSE没有持续抑制IFN-γ-mediated immunoproteasome诱导表达后24小时(补充图S5a),我们注意到减毒诱导类MHC分子(HLA-ABC)在流式细胞术分析(补充图S5b)。
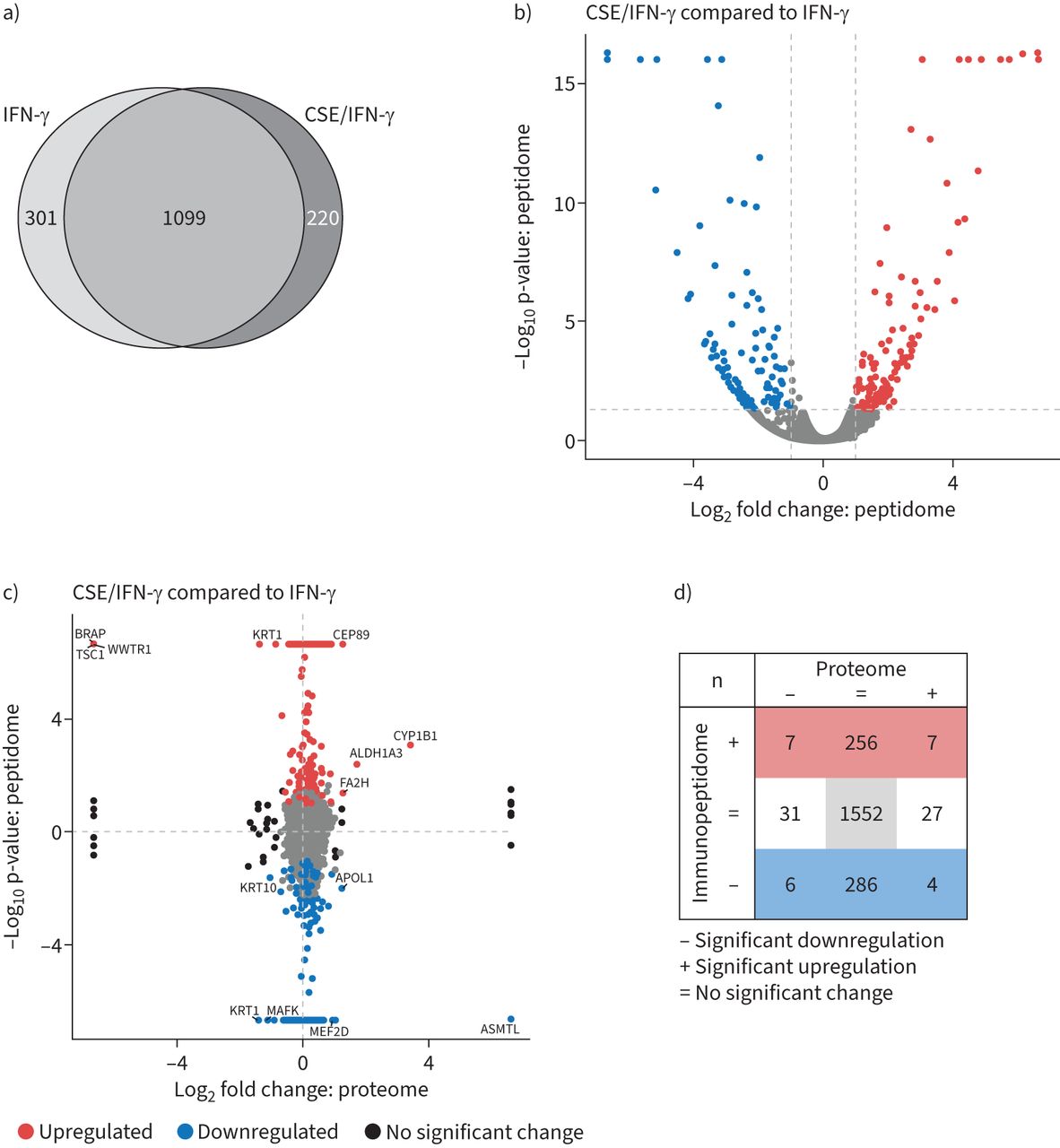
immunopeptidome分析,我们首先确定多肽的数量满足HLA-binding的要求。概述了严格的选择标准(见补充的方法详情),我们发现在400年和1500年之间每治疗HLA-bound肽(补充图S6a)。识别抗原肽的数量高于IFN-γ-treated细胞,这是符合以前的观测(35]。CSE-treatment的细胞没有严重变化的HLA-peptides基线也在IFN-γ-treatment (补充图S6a)。肽来源于不同的HLA分布类我等位基因是影响IFN-γ,如图所示(前36),但不是由CSE治疗(补充图S6b)。作为IFN-γ治疗诱导最明显的变化在整个抗原,我们集中分析CSE治疗的效果在IFN-γ-induced immunopeptidome。我们发现220新CSE后我抗原肽MHC类/ IFN-γ-treatment IFN-γ相比所呈现的维恩(图5一个)和火山地块(图5 b)。新抗原肽来自多个不同本体术语相关的蛋白质和各种细胞本地化和分子功能没有浓缩为特定通路(数据未显示)。此外,301年IFN-γ-treated肽可再生产地确定细胞CSE / IFN-γ-treatment(后普遍较低图5一个和b)。这也源于蛋白质不同的本体,功能和途径。
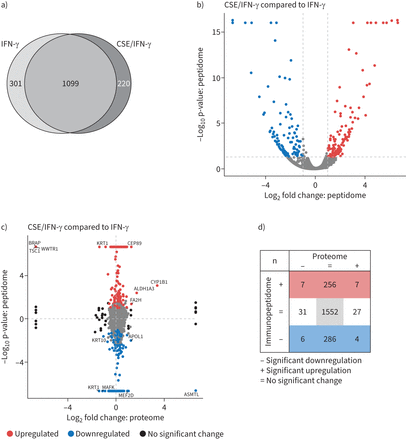
香烟烟雾提取物(CSE)对炎症主要组织相容性复合体(MHC)我immunopeptidome类。)维恩图的重叠和独特的MHC类我肽中标识immunopeptidome A549细胞(绑定等级≤2%,信心得分≥4.2,n = 3)。细胞被刺激了75 U·毫升−1干扰素(IFN) -γ24 h或预处理的20% CSE 48 h,然后用75 U·co-stimulated毫升−1IFN-γ过去24小时。b)火山的情节认定MHC类我肽)。显著调节肽CSE IFN-γ-treated细胞单独IFN-γ治疗中所示红色相比,显著下调肽用蓝色表示。冲水平线表示意义阈值为0.05。这两个虚线竖线表示日志2−1和1的相对丰度阈值。c)丰富immunopeptidome中标识肽和蛋白质的比较中发现的蛋白质组CSE IFN-γ-treated细胞相比IFN-γ单独治疗。显著调节我肽MHC类描述为红色,而大幅下调MHC类我肽是蓝色的。蛋白质显著调节蛋白质组没有显著变化peptidome显示为黑色。肽显著调节蛋白质组和immunopeptidome与他们的基因符号标记。d)枚举的显著调节肽c)。
接下来,我们研究如何在细胞蛋白质丰度差异转化为改变MHC抗原类我表示对CSE和IFN-γ治疗相结合。因此,我们执行额外的猎枪蛋白质组学分析为每个治疗条件,然后绘制日志2倍的变化我类MHC抗原肽y轴到日志中2倍改变蛋白质的丰度在x轴上每一对治疗(图5 c和补充图S6c)。CSE / IFN-γ-treatment调节七抗原肽由于蛋白水平升高(右上象限,图5 c和d)。提出了七个抗原肽蛋白质的表达下调相比IFN-γ-treated控件(左上象限,图5 c和d)。调节抗原肽大部分是来自蛋白质,没有改变表达式的CSE / IFN-γ相比只有IFN-γ治疗(图5 c和d)。充足比率immunopeptidome和蛋白质组的变化不太明显,当我们相比CSE-treated细胞治疗细胞表明最初的蛋白质组改造主要由IFN-γ驱动而不是CSE治疗(补充图S6c)。我们得出结论,CSE形状与IFN-γimmunopeptidome A549细胞在炎症刺激,但只有一个小对稳态蛋白质组的影响。这种改造的炎性抗原肽曲目由香烟烟雾影响CD8的潜力+病毒感染t细胞激活和适应性免疫反应。
慢性阻塞性肺病患者循环IAV-specific CD8数目减少+t细胞
考虑到病毒特异性CD8降低的结果+t细胞激活的香烟烟雾,我们推断,延长暴露于香烟烟雾吸烟者和慢性阻塞性肺病患者有助于激活受损的抗病毒的适应性免疫反应。这可能是检测病毒特异性CD8数目减少+在外周血t细胞。我们测试了这个概念通过分析IAV-specific CD8的数量+t细胞在孤立的外周血单核细胞(PBMCs),比较不吸烟者ever-smokers(研究对象1)(补充表S1为研究队列)和比较严重的慢性阻塞性肺病患者肺部健康疾病和哮喘控制(研究队列2)。所有慢性阻塞性肺病患者曾经吸烟持续时间从10到50久(表1)。确定IAV-specific t细胞的数量,我们使用了四聚物技术和专注于immunodominant HLA-A2-restricted IAV M158 - 66抗原决定基述[37局限于hla a2。hla a2与M1四聚体加载58 - 66结合具体IAV-specific CD8+t淋巴球,然后可以量化通过流式细胞术(补充图S7对控制策略和控制)。因为这IAV hla a2抗原决定基是要受到限制的,我们的研究参与者首先选择hla a2表达式(表1和补充表S1)。在我们的第一个研究对象、IAV-specific CD8+t细胞(CD3总量的百分比+细胞)不吸烟者之间没有显著差异(n = 7,包括一个水烟吸烟者)和ever-smokers (n = 7) (图6)。CD3的比例之间没有相关性+CD8+IAV-tetramer+t细胞和久(数据没有显示)。然而,在慢性阻塞性肺病病人群体,CD3的绝对数量+CD8+IAV-tetramer+严重的慢性阻塞性肺病患者的t细胞是低比lung-healthy控制和比哮喘患者(图6 b,补充图S8a)。同样,IAV-specific CD8的频率+t细胞在慢性阻塞性肺病患者显著降低的时候,CD3的总数+细胞出现在这些患者PBMC样本(图6 c,补充图S8b)。这些数据支持这一概念,慢性阻塞性肺病病人抗病毒CD8受损+t细胞反应(17,18]。然而,少量的研究参与者及其异构的吸烟习惯(补充表S1)限制的结论和需要进一步分析。

测定甲型流感病毒(IAV特殊CD8)+t细胞的血液lung-healthy控制和慢性阻塞性肺病患者。CD3的百分比+CD8+IAV-tetramer(春节)+t细胞内所有CD3的分数+细胞在外周血单核细胞(PBMCs) lung-healthy控制那些不吸烟者(n = 7,包括一个水烟吸烟者)与烟或当前吸烟者(n = 7)。b) CD3的绝对数量+CD8+IAV-tetramer+t细胞(细胞·μL−1)孤立PBMCs COPD患者(n = 9)和年龄lung-healthy控制(n = 10)。临床特点中可以找到表1。c) CD3的百分比+CD8+IAV-tetramer+t细胞内所有CD3的分数+细胞PBMCs肺健康对照组(n = 9)和慢性阻塞性肺病患者(n = 10)。数据作为中位数(四分位范围);意义测试使用Mann-Whitney紫外线测试。* *:*:p < 0.05, p < 0.01。
讨论
在这项研究中,我们表明,吸烟会损害细胞因子,占据upregulation immunoproteasome /类MHC抗原表示机械associates的抗病毒CD8的激活+t细胞。从我们使用不同的模型系统在体外鼠标的细胞系,体外肺的文化,在活的有机体内老鼠实验的主要人体细胞和临床相关的病毒的测试,即。IAV HRV-16。此外,质谱仪分析方面的主要改造基础immunopeptidome炎症类MHC抗原曲目我吸烟。这个概念,香烟烟雾干扰有效抗病毒CD8+t细胞免疫被发现进一步证实了严重的慢性阻塞性肺病患者吸烟史的流感特定外围CD8降低了数字+t细胞与肺健康对照组和哮喘病人。
香烟烟雾干扰IFN-mediated抗病毒免疫反应
而我们之前的研究分析了香烟烟雾对immunoproteasome函数的影响免疫细胞在肺,肺组织(26,38)和外周血细胞(39),在这里,我们专注于香烟烟雾对细胞因子的影响,virus-mediated感应多发地immunoproteasome的细胞。我们的数据符合和扩展先前的研究报告减少响应性上皮细胞干扰素或病毒暴露在香烟烟雾提取物(40- - - - - -42]。此外,我们的研究结果支持最近的研究在香烟烟雾暴露SARS-CoV-2感染呼吸道上皮细胞(43),体外人类肺文化(31日]。虽然这些研究的重点是在抗病毒先天免疫的抑制CSE以及CSE干扰素干扰信号(44,45),我们的数据显示CSE-induced障碍之间的关联在感染细胞的干扰素反应和适应性CD8+t细胞激活通过immunoproteasome /类MHC抗原表达机械。我们从而提供一个机械链接之前报道CD8的障碍+香烟smoke-exposed小鼠t细胞活化在病毒感染(14,46]。我们观察到不同的香烟烟雾对个人immunoproteasomal催化亚基的表达。底层机制需要进一步调查。然而,赞赏的催化亚基immunoproteasome炎症和压力条件下不同规范(21,47]。为此,我们最近演示了在一个大的人口基数,催化亚基的活性蛋白酶体的外周血细胞不同监管根据性别与年龄和慢性炎症性疾病48]。总的来说,immunoproteasome函数不仅是由催化亚基的表达,也被他们的蛋白水解活性,由香烟烟雾[受损26,29日,38]。值得注意的是,吸烟不仅抑制细胞因子和virus-mediated感应immunoproteasome的多个模型包括不同的病毒,而且中和病毒诱导upregulation MHC类的分子,这符合先前的报道(26,49]。immunopeptidomic分析揭示了一个额外的监管层烟,诱导炎症MHC类的我immunopeptidome质的变化。我们观察到> 200小说抗原肽在CSE / IFN-γco-treated IFN-γ相比在几个抗原刺激细胞失去了对CSE治疗。这些抗原来源于蛋白质跨多个细胞通路而不是丰富为特定分子功能。这些多肽蛋白质水平上没有监管。综上所述,我们在这里证明香烟烟雾干扰MHCⅰ类介导的抗原表达在多个层面上从减少immunoproteasome感应,改变我upregulation MHC抗原表位的生成和减少类。所有这些机制增加CD8降低+t细胞活化在这项研究中观测到的。我们的数据从而提供重要的机械的见解如何吸烟会损害CD8+t细胞免疫反应,阻碍病毒清除。
慢性阻塞性肺病患者的抗病毒免疫缺陷
我们的在体外数据显示有缺陷的MHC类我演示和抗病毒CD8抗原+t细胞激活支持我们的观测频率的流感特定外围CD8降低+t细胞在慢性阻塞性肺病患者与健康对照组和哮喘患者。我们的数据符合从2005年的一项研究报告RSV-specific CD8水平较低+t细胞在慢性阻塞性肺病患者的血50]。IAV-specific和总CD8的功能+t细胞表面表达在慢性阻塞性肺病患者评估的脱粒标记CD107a胞内IFN-γ染色并没有改变(数据未显示)。这一发现发表的结果是一致的报告类似的健康COPD-derived CD8细胞因子的生产+t细胞后在体外疫苗刺激(51),但与观察CD8炎症性能力的提升+慢性阻塞性肺病患者t细胞激活的时候在体外(52]。相互矛盾的数据进一步报道有关数字,循环和lung-resident CD8的表型和功能+稳定的慢性阻塞性肺病患者的t细胞(53,54]。然而,最近的一项荟萃分析这个话题似乎支持高浓度的外围CD8的概念+t细胞在慢性阻塞性肺病,缺乏完整的功能(53]。对于lung-resident CD8+t细胞,upregulation程序性细胞死亡蛋白1 (PD)和减少功能报道(55]:威尔金森实验室证明体外感染的COPD肺组织IAV调节PD-1 CD8+t细胞来源于慢性阻塞性肺病组织更强烈而控制组织伴随着减少脱粒这些细胞的能力。Bitonet al。(56报道肺肿瘤居民CD8疲惫+伴随慢性阻塞性肺病患者的t细胞更普遍的全球倡议对慢性阻塞性肺疾病阶段II + III。一个优雅的老鼠实验表明,香烟烟雾诱发肺气肿早期细胞毒性CD8功能受损+t细胞的反应对肺移植癌细胞(15]。这些观察结果提出有趣的问题是否无效的清除有缺陷的CD8的病毒感染+t细胞免疫可能不仅有助于增加慢性阻塞性肺病患者病毒发作的易感性,但也推动疾病发展潜在的病毒感染和香烟添加增效剂在慢性阻塞性肺病慢性炎症和组织损伤,正如前面提出的(3]。
力量和本研究的局限性
在这项研究中,我们分析了香烟烟雾对几个组件的影响我类MHC抗原表达的复杂的过程,即。改变virus-mediated immunoproteasome感应,减少类MHC抗原决定我表面表达和一代的小说。为此,我们使用在体外,体外和在活的有机体内模型允许我们获得概念的证据单一组件上的香烟烟雾的影响。最有可能的是激活CD8降低+t细胞通过吸烟涉及的损害整个immunoproteasome /类MHC抗原表达机械。我们的研究仅限于使用单个HLA-A2-restricted IAV抗原决定基,即M158 - 66抗原。我们已经关注这一抗原决定基众所周知,这种抗原的生成取决于immunoproteasome[的催化活性33,57]。我们可以因此CD8联系起来+t细胞激活immunoproteasome的活动。当有多个其他知名IAV抗原和相应的CD8+可用(t细胞克隆32),还不知道这些病毒抗原的生成取决于immunoproteasome。我们的四聚物分析关注的另一个原因是HLA-A2-restricted IAV抗原决定基是hla a2的最普遍的基因家族在所有少数民族人口58]。尽管有这些限制,我们的数据提供了重要的机械的见解香烟如何阻碍抗病毒CD8+t细胞反应。不过,研究参与者的数量在我们两个研究武器相当小。然而,合并肺健康控制和一种哮喘疾病对照组加强我们的结论。重要的是,我们的研究提供了概念性的证据改变类MHC抗原表达在慢性阻塞性肺病疾病恶化,这个话题已经超到目前为止。
补充材料
可共享的PDF
确认
我们请承认IAV的慷慨礼物应变PR8从Susanne哈罗德(李比希大学卓越集群心肺研究所(CPI),吉森大学和马尔堡肺中心(UGMLC),德国的肺癌研究中心(DZL) Gießen,德国)和提供IAV-specific t细胞杂种细胞克隆4 va1由大卫·堪(克利夫兰凯斯西储大学哦,美国)。此外,我们感谢专家的支持比阿特丽克斯引导(研究中心Borstel,莱布尼茨肺中心气道北方研究中心(ARCN),德国的肺癌研究中心(DZL) Borstel,德国)。我们感谢美国国立卫生研究院四聚物核心设备(合同号75 n93020d00005)提供IAV四聚体。图形与BioRender文摘生成。我们感激地承认人类生物材料的供应和临床数据CPC-M bioArchive及其合作伙伴在Asklepios生物Gauting, LMU医院和Ludwig-Maximilians-Universitat慕尼黑。我们感激的优秀的技术援助Frauke瘦身,Gesine骑和基督教Rosero Borstel(研究中心)。我们也感谢Margrit Kernbach和斯蒂芬妮福克斯BioMaterialBank北(Borstel、德国),优秀的技术援助,和研究中心研究中心Borstel对他们的帮助。我们感谢病人和他们的家属以及健康献血者的支持。
脚注
x作者贡献:即Kammerl、陈、王、美国Meiners负责概念和设计的研究;m .私生子K.I. Gaede, k . Milger J.S.二李,j .品牌和j·贝赫(临床)提供样品和试剂;j·陈,x,海恩斯,a . Schmalen M.G. Stoleriu, m . Nakayama m·布埃诺j .品牌,m·沃尔夫·h·马,a . Dmitrieva j·诺瓦克和即Kammerl进行实验;j·陈,x,海恩斯,a . Schmalen h .妈,j . Behrends t . Goldmann c.a Staab-Weijnitz, A.L.•莫拉·m·布埃诺J.S.二李,m·沃尔夫张h, s . Krauss-Etschmann h·阿德勒,克里·豪t而,e . Noessner c.a Deeg, a . Moosmann即Kammerl和美国Meiners分析数据;即Kammerl A.L.莫拉,e . Noessner a . Moosmann h·阿德勒,t . Goldmann Behrends和美国Meiners解释结果;即Kammerl、x Wang Meiners和j·陈准备表和数据;即Kammerl, x, s . Meiners a Schmalen和j·陈起草的手稿;王即Kammerl、x和美国Meiners编辑和修订后的手稿;所有作者批准了最终版本。
利益冲突:h . Ma报告支持德国肺癌研究中心目前的手稿(DZL)。m . Nakayama报道海外格兰特Uehara纪念基金会(日本)和海外拨款志贺医学大学外提交的工作。A.L.莫拉报告支持目前的手稿来自国家卫生研究院(NIH U01 HL1455550-01 NIH NHLBI R01 HL149825)。J.S.二李与詹森研发、参与临床审判委员会报告在提交工作。美国Krauss-Etschmann报告支持德国肺癌研究中心目前的手稿。k . Milger讲座报告咨询费和阿斯利康的谢礼,葛兰素史克,詹森,诺华公司和赛诺菲,在提交工作。c.a Staab-Weijnitz报告从亥姆霍兹联合会支持目前的手稿,德国肺癌研究中心(DZL)和德意志Forschungsgemeinschaft GRK2338 (DFG)在研究培训组。K.I. Gaede报告支持目前的手稿研究中心Borstel -莱布尼兹肺中心BioMaterialBank北气道北方研究中心,德国肺癌研究中心(DZL) PopGen 2.0网络(P2N)。K.I. Gaede还持有领导角色作为TMF的董事会成员(www.tmf-ev.de),在提交工作。从人即Kammerl报告支持目前的手稿(短期奖学金)。 All other authors have no potential conflicts of interest to declare.
支持声明:资金支持的研究从医学院LMU s·海恩斯(FoFoLe程序),欧洲呼吸协会(ERS)(即Kammerl短期奖学金),校内的亥姆霍兹慕尼黑中心的资金,和德国联邦教育和研究,德国(BMBF)授予EXASENS (13 n13856) s Krau188bet官网地址ss-Etschmann m·沃尔夫(资金)。j·陈是由中国国家自然科学基金(81600063)。h .支持中国博士后科学基金会(2022 m720916)。m . Nakayama Uehara纪念基金会的支持下,志贺大学医学科学。美国Meiners是通过个人由莱布尼茨基金会拨款资助。资金信息,本文已沉积的Crossref资助者注册表。
- 收到了2022年7月15日。
- 接受2023年5月24日。
- 版权©2023年作者。
这个版本分布在创作共用署名非商业性许可证的条款4.0。商业生殖权利和权限接触权限在}{ersnet.org











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
![Cigarette smoke impairs virus-mediated induction of the immunoproteasome and major histocompatibility complex (MHC) class I in human epithelial cells. a) Low molecular weight protein (LMP)2 and LMP7 expression in primary human bronchial epithelial cells (pBAECs) from three different donors that had been cultured at air–liquid interface conditions with or without cigarette smoke extract (CSE) in their medium [30] and infected with influenza A virus (IAV) for 3 days [60]. Densitometric analysis of LMP2 and LMP7 expression normalised to β-actin with untreated group set to 1. b) pBAECs from two male healthy donors were infected with human rhinovirus (HRV)-16 for 2 days after exposure to two cigarettes per day for 14 days during the air–liquid interface differentiation phase. mRNA levels for PSMB8 (LMP7), PSMB9 (LMP2) and PSMB10 (multicatalytic endopeptidase complex subunit (MECL)-1) and IFNB1 (interferon-β) were determined using reverse transcriptase quantitative PCR and related to housekeeping gene expression (HPRT and RPL32). Data are shown as four replicates per donor (mix of two technical and two independent cultures) with mean±sem. c) Human leukocyte antigen (HLA)-ABC surface expression of A549 cells treated with 20% CSE for 24 h and infected with IAV (multiplicity of infection 1) for 24 h. Fluorescence intensity is shown as mean±sem of three independent experiments. Ctrl: control; Iso: isotype; FITC: fluorescein isothiocyanate; ΔMFI: change in mean fluorescence intensity. One-way ANOVA with Bonferroni post-test. *: p<0.05, **: p<0.01, ***: p<0.001.](http://www.qdcxjkg.com/content/erj/62/2/2201374/F4.large.jpg?width=800&height=600&carousel=1){kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}