





摘要
CXCL10刺激肥大细胞浸润到气道平滑肌束,从而激活细胞因子分泌和气道平滑肌细胞(ASMC)增殖。富马酸二甲酯(DMF)通过血红素加氧酶(HO)-1减少淋巴细胞分泌细胞因子和ASMC增殖。因此,我们研究了DMF对人ASMCs肿瘤坏死因子(TNF)-α和干扰素(IFN)-γ诱导的CXCL10分泌的抑制作用。
在用IFN-γ和/或TNF-α刺激细胞之前,用DMF和/或氟替卡松和/或谷胱甘肽乙酯预培养人原代asmc。
DMF抑制CXCL10的分泌并增加HO-1的水平,p38丝裂原活化蛋白激酶(MAPK)的抑制降低了DMF依赖性的HO-1的表达。通过HO-1小干扰RNA (small interfering RNA, siRNA)预处理,消除DMF对CXCL10分泌的影响。补充谷胱甘肽逆转了DMF对CXCL10分泌和p38 MAPK磷酸化的所有影响。重要的是,DMF与氟替卡松联合使用进一步减少了CXCL10的分泌。此外,DMF抑制IFN-γ诱导的CXCL10分泌。这种效应可以通过谷胱甘肽补充或HO-1 siRNA预处理来补偿。此外,DMF可减少TNF-α诱导的粒细胞集落刺激因子(G-CSF)的分泌,但对INF-γ诱导的G-CSF分泌无影响。
在人类初级asmc中,DMF通过降低细胞谷胱甘肽水平和激活p38 MAPK和HO-1来抑制CXCL10的分泌。因此,DMF可能通过糖皮质激素非依赖性途径减轻哮喘气道炎症。
哮喘是一种慢性气道炎症性疾病,其特征是气道高反应性、支气管收缩增加和气道壁厚度增加[1].哮喘中的气道炎症涉及组织形成细胞,如上皮细胞、成纤维细胞和气道平滑肌细胞(ASMCs),以及循环免疫细胞,主要是肥大细胞、t细胞和嗜酸性粒细胞[2].活化的asmc在肿瘤坏死因子(TNF)-α等细胞因子的刺激下分泌多种促炎因子,从而将更多的炎症细胞招募到炎症区域[3.].
CXCL10是一种有效的肥大细胞趋化有助于哮喘的发病机理[4].为CXCL10的哮喘的病理贡献支持来自观察,即从激活的哮喘组ASMCs收集细胞培养基诱导的人肺肥大细胞的趋化性[5].当CXCL10中和或减少这种效应当其受体CXCR3被阻断[6].此外,哮喘患者asmc分泌的CXCL10比健康对照组细胞分泌的CXCL10更多[7].这与哮喘患者气道平滑肌束中活跃的肥大细胞数量增加有关。此外,CXCL10分泌和肥大细胞数量与气道高反应性相关[4].
吸入糖皮质激素是治疗哮喘最常用的抗炎药物[7].然而,在10%的哮喘患者中,气道炎症没有得到常规治疗的良好控制,这些患者占哮喘医疗保健总费用的50% [8].在细胞培养模型中,已经表明,炎性干扰素(IFN)-γ的信号传导途径上调的表达CXCL10由主动脉平滑肌细胞,且这是类固醇抗性[9].因此,需要新的治疗方案来控制哮喘患者的气道炎症。
据坊间传言,dimethylfumarate (DMF;也被称为BG00012)已被报道可以减轻哮喘症状,并改善患有哮喘的银屑病患者的生活质量。在体外,DMF改变了细胞内还原型谷胱甘肽(GSH)的代谢,从而降低了促炎细胞因子的表达[10.,11.].在人外周血单个核细胞中,DMF清除细胞内GSH,诱导血氧合酶(HO)-1,抑制白细胞介素(IL)-12和IFN-γ的分泌。当谷胱甘肽被补充时,这些作用被削弱了[10.].关于DMF在多发性硬化症中的有益作用,它被认为是有效的通过细胞保护转录因子Nrf-2,在谷胱甘肽缺失时被激活[9].在人asmc中,我们发现DMF抑制TNF-α诱导的IL-6、eotaxin和RANTES(调节活化、t细胞表达和分泌)的分泌[12.].此外,我们观察到DMF通过HO-1抑制血小板衍生生长因子(PDGF)- bb诱导的ASMC增殖[13.].DMF与临床长期安全性一起,这些有益的作用,使其成为慢性炎症性肺疾病的治疗[有趣的药物14.].
在这项研究中,我们研究了DMF对人ASMCs HO-1表达的影响,以及它对TNF-α和/或IFN-γ诱导的CXCL10和粒细胞集落刺激因子(G-CSF)分泌的作用。此外,我们研究了丝裂原活化蛋白激酶(MAPK)激活在dmf刺激HO-1表达的背景下的作用。我们还评估了谷胱甘肽补充对这一信号系统的影响,以及DMF可能的类固醇保存特性。
材料和方法
肺组织标本
本研究中使用的所有标本均来自巴塞尔大学医院(瑞士巴塞尔)内科,获得了当地伦理委员会的批准和所有患者的书面同意。
人asmc的分离和培养
人类原代asmc是从未使用的肺移植获得的健康肺组织的支气管中分离和生长的,并如前所述建立[15.].主动脉平滑肌细胞是在RPMI-1640(ThermoTrace,墨尔本,澳大利亚),补充了5%(V / V)热灭活的胎牛血清(FBS),1×最低必需培养基维生素混合物,100U·L生长−1青霉素,100μg·mL的−1链霉素、0.25μg·毫升−1两性霉素B(所有GIBCO / BRL,墨尔本,澳大利亚),25mM的哌嗪羟乙基乙烷磺酸和2mMl-谷氨酰胺(均为ThermoTrace),在37°C、5%二氧化碳的湿化气氛中。第5代和第8代之间使用asmc。
药物制剂
自Sigma(Buchs,瑞士)获得所有的化学品。DMF(0.1-100μM),SB203580(10μM),氟替卡松(0.1-10μM),氯高铁血红素(1-10μM)和原卟啉钴(2-20μM)溶解于二甲亚砜(DMSO),并稀释至在无血清培养基中的所需浓度。谷胱甘肽乙酯(GSH-OET)溶解至1mM的在无血清培养基中的最终浓度。DMF浓度的细胞毒性功效并不显著,如先前报道[13.].
asmc分泌CXCL10
asmc种植融合和生长介质被low-serum取代培养基(0.1%的边后卫)24 h,在DMF添加1 h。细胞被刺激与肿瘤坏死因子-α和/或干扰素-γ(研发系统,明尼阿波利斯,美国)和细胞培养基是24或48小时后收集样品。CXCL10和G-CSF蛋白水平的测定采用ELISA试剂盒按照制造商说明(ELISA Duo Set;研发系统)。
小干扰RNA转染
细胞播种到12孔培养板(1×105细胞·毫升−1),并在RPMI中增长到70%汇合(10%胎牛血清)。用无fbs的RPMI洗涤细胞,然后用5 μL siRNA转染试剂瞬时转染10 μM小干扰RNA(靶向HO-1或各自的对照siRNA)(所有Santa Cruz Biotechnology, Heidelberg, Germany)。单独用转染试剂孵育的细胞作为阴性对照。siRNA处理6小时后,用生长培养基(RPMI, 10%胎牛血清和1×抗生素)刺激细胞20小时。PBS冲洗细胞,用DMF (50 μM)孵育1小时,TNF-α或IFN-γ(5或10 ng·mL)−1)补充道。24 h后采集细胞培养液样品,ELISA检测CXCL10蛋白含量。
HO-1表达与MAPK激活
asmc培养至融合,然后血清饥饿24小时(0.1%胎牛血清)。在用TNF-α刺激细胞之前,用单一药物或药物组合预处理细胞(1 h)。在不同时间点(0、5、10、15、30、60 min)收集细胞总裂解液,通过免疫印迹法检测MAPKs的表达和激活情况。24 h后检测HO-1的表达。
免疫印迹
蛋白质提取物用十二烷基硫酸钠-聚丙烯酰胺凝胶电泳进行大小分级,并如前所述转移到硝基纤维素膜上[13.].Ponceau染色证实蛋白转移。膜是孵化阻断缓冲区(5% (w / v)脱脂奶粉Tris-buffered盐水含0.1%渐变20)在室温下1 h,然后被孵化主要抗体特定要么p38,磷酸化p38,细胞外signal-regulated激酶(ERK) 1/2,磷酸化ERK1/2, c-Jun n端激酶(物),磷酸化JNK (all Cell Signaling Technology, Beverly, MA, USA), HO-1 (Calbiochem, Luzern, Switzerland)或α-微管蛋白(Santa Cruz Biotech)。用1∶2 000 - 1∶4万稀释的辣根过氧化物酶偶联免疫球蛋白G(抗兔IgG或抗小鼠IgG;圣克鲁斯生物技术)。通过增强化学发光显示蛋白质条带(Pierce Biotechnology, Rockford, IL, USA)。
数据分析
一式三份或每个实验重复结果取平均的每个主ASMC线,并从这些数据中,平均值±扫描电镜计算了。采用单因素方差分析进行统计分析。p值由Bonferroni方法修正,分别在图中显示。
结果
DMF抑制ASMCs TNF-α诱导的CXCL10分泌
用TNF-α (10 ng·mL)刺激ASMC−1)或静刺激24、48 h后测定细胞培养液中CXCL10的水平。所示图1一个,未受刺激的ASMCs释放低水平的CXCL10(平均±扫描电镜69.9±27.1 PG·毫升−1),而TNF-α刺激CXCL10在24 h显著上调(1,044±284 pg·mL)−1)和48小时(1382±316皮克·毫升−1).DMF (1-100 μM)预培养剂量依赖性地减少TNF-α诱导的CXCL10分泌,而DMF单独对未刺激asmc无影响(图1 b).与预期一样,氟替卡松(0.1-10 μM)可抑制TNF-α诱导的CXCL10分泌,与DMF (10 μM)联用时,其作用比单独用药更强(图1 c).
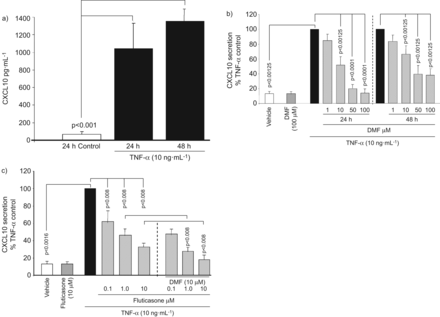
富马酸二甲基酯(DMF)抑制肿瘤坏死因子(TNF)-α诱导的CXCL10分泌,并由糖皮质激素支持。a)用TNF-α (10 ng·mL)刺激细胞−1)或不处理24或48 h。ELISA检测细胞培养液中CXCL10。数据表示为平均值±扫描电镜对10个独立细胞系进行3次重复实验,并与未受刺激的细胞进行配对t检验,计算p值。b)气道平滑肌细胞(n=8)在TNF-α刺激前用DMF或0.1%二甲基亚砜处理1小时。在24和48小时用ELISA法测量CXCL10分泌。数据以TNF-α单独处理CXCL10产生细胞的百分比表示,p值通过单因素方差分析计算。c)采用氟替卡松单独或联合DMF治疗asmc (n=6) 24小时,ELISA检测CXCL10分泌情况。数据表示为平均值±扫描电镜对6个独立ASMC品系进行三次重复实验,采用方差分析计算p值,并用Bonferroni方法进行校正。
通过HO-1 DMF抑制CXCL10分泌由主动脉平滑肌细胞
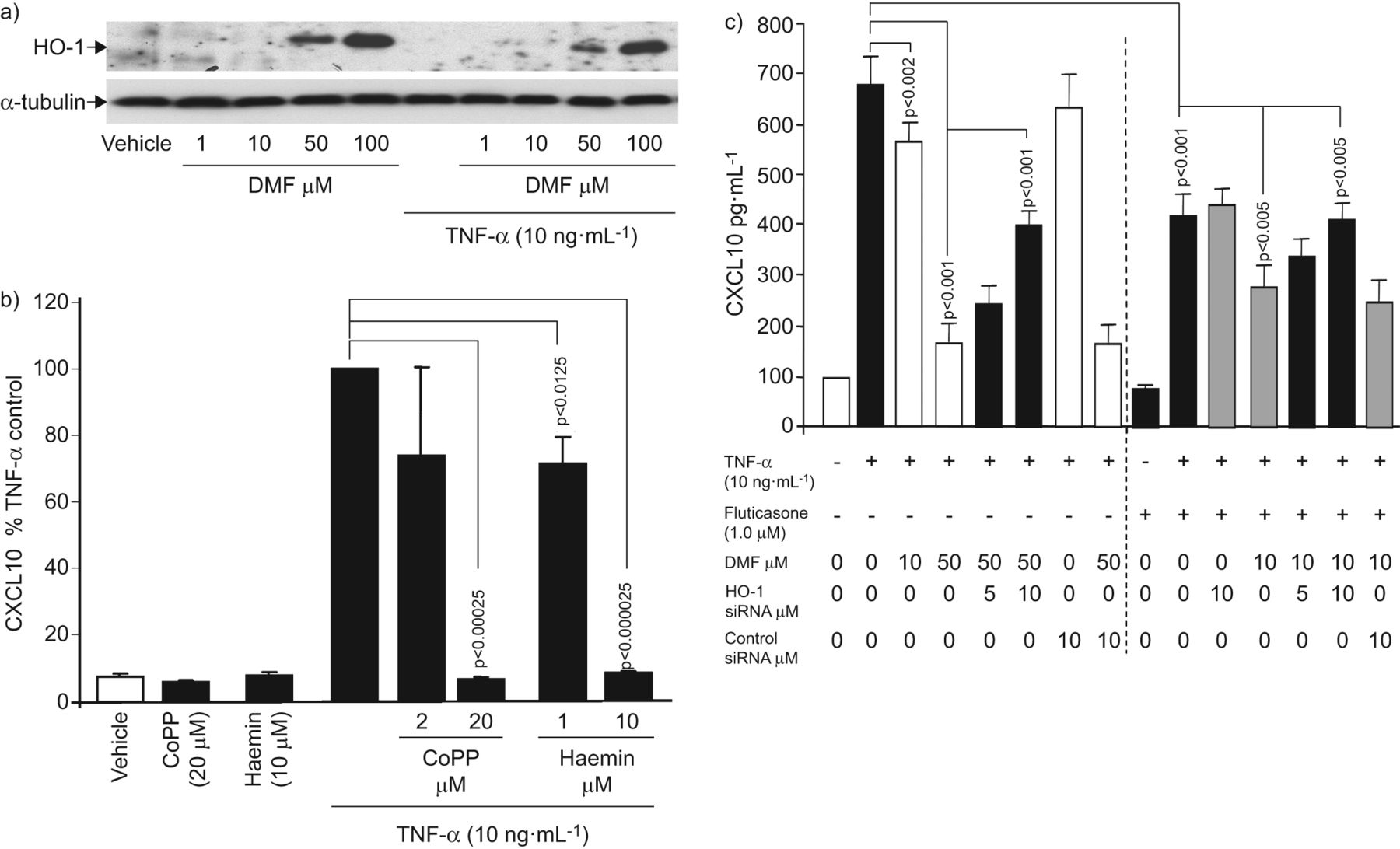
用免疫印迹法检测HO-1的表达。DMF诱导HO-1表达呈剂量依赖性,TNF-α既不诱导也不改变DMF诱导的HO-1表达(图2一个).为了证实HO-1对CXCL10分泌的抑制作用,在用TNF-α刺激asmc之前,用haemin (1 - 10 μM)或钴原卟啉(2-20 μM)预培养1小时。这两种HO-1诱导剂剂量依赖性地减少了asmc中TNF-α诱导的CXCL10分泌,但对未刺激的细胞无影响(图2 b).
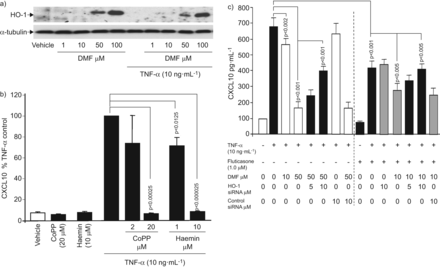
血红素加氧酶(HO)-1介导二甲酯的抑制效果(DMF)上通过CXCL10气道平滑肌细胞(主动脉平滑肌细胞)的分泌。a)该DMF的代表性免疫印迹剂量依赖性的HO-1表达在第24小时通过主动脉平滑肌细胞(N = 3)。类似的结果在另外五个细胞系获得。b)在HO-1的诱导原卟啉钴(COPP)和氯高铁血红素抑制肿瘤坏死因子(TNF)在24小时-α诱导CXCL10分泌。数据表示为平均值±扫描电镜用四种独立细胞系进行三次重复实验,用单因素方差分析计算经TNF-α处理的细胞产生CXCL10的百分比和p值。c)用ho -1特异性小干扰RNA (siRNA)转染asmc (24 h),然后用DMF (50 μM)或氟替卡松联合DMF处理1 h,然后用TNF-α (10 ng·mL)处理−1)刺激24 h, CXCL10分泌为平均值±扫描电镜在未处理细胞中10次独立的实验,和p值的该百分比是通过ANOVA计算和Bonferroni方法进行校正。
HO-1在DMF对TNF-α诱导CXCL10分泌抑制效应的作用也进行了研究中已被暂时以DMF和TNF-α治疗转染HO-1的siRNA前主动脉平滑肌细胞。所示图2 c结果表明,50 μM DMF对CXCL10分泌的抑制作用可被HO-1 siRNA剂量依赖性地抵消(p<0.05),而对照组siRNA则无此作用。重要的是,单独氟替卡松对TNF-α诱导的CXCL10的抑制作用不受HO-1 siRNA的影响,与联合药物处理时,抵消了DMF的作用(图2 c).
DMF的增加和延长TNF-α诱导的p38蛋白激酶磷酸化
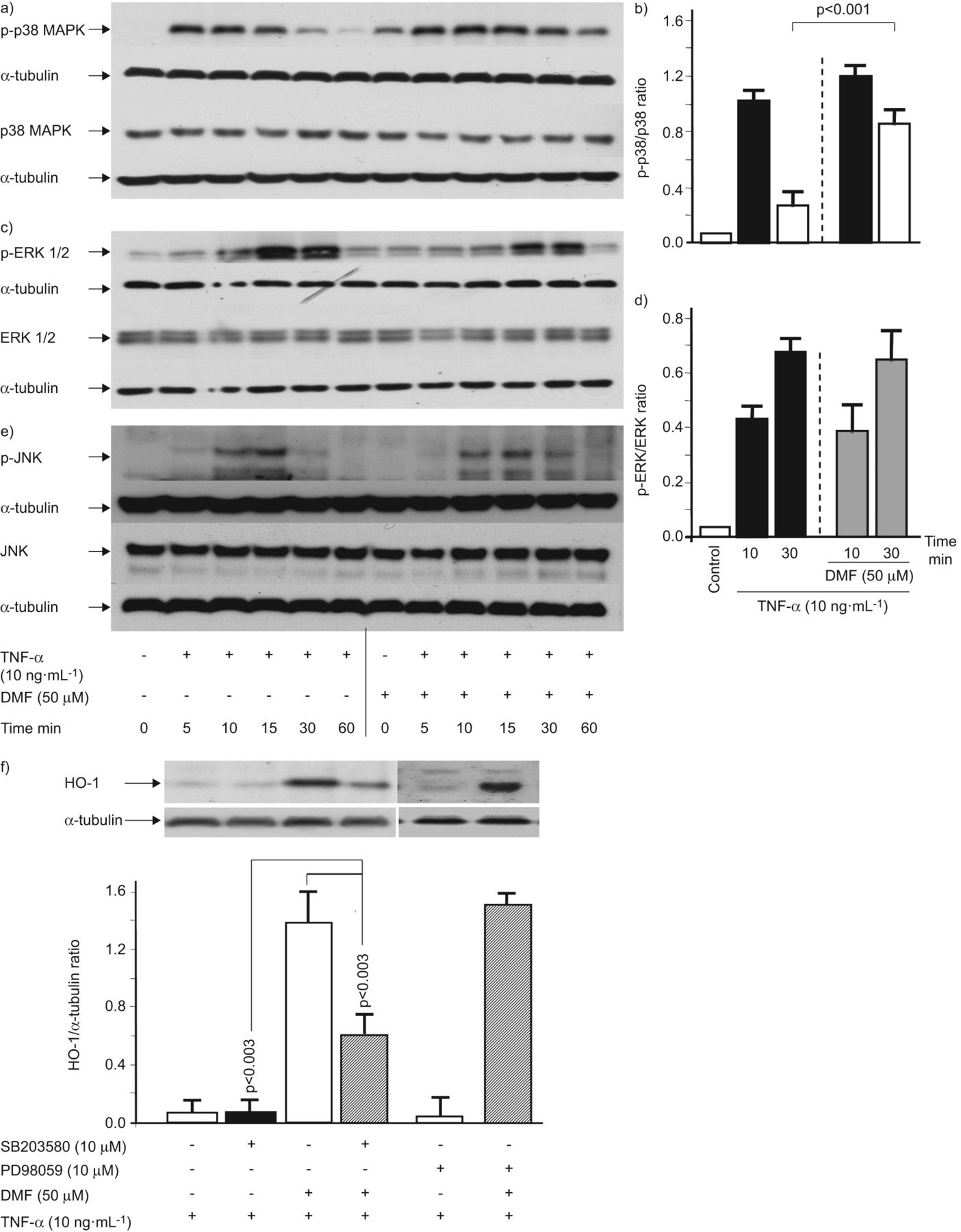
肿瘤坏死因子-α(10 ng·毫升−1)在5分钟内强烈诱导p38 MAPK磷酸化,随后下降,在60分钟时达到基线水平(无花果。图3a和b).DMF (50 μM)单独激活p38 MAPK磷酸化,当与TNF-α联合使用时,它增加并延长p38 MAPK磷酸化(无花果。图3a和b).任何处理均不影响p38 MAPK总表达(无花果。图3a和b).TNF-α诱导的ERK1 / 2 MAPK和JNK的磷酸化,这两者都不是受DMF(图3汉英).任何处理均未观察到总ERK1/2 MAPK或总JNK水平的变化(图3汉英).
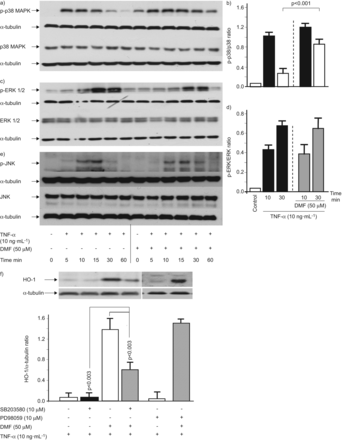
富马酸二甲酯(DMF)通过气道平滑肌细胞激活p38丝裂原活化蛋白激酶(MAPK),其介导血红素加氧酶(HO)-1表达的磷酸化。一)p38蛋白(P-p38蛋白)的磷酸化的动力学的代表性免疫印迹,和b)通过定量肿瘤坏死因子(TNF)-α的DMF的存在和不存在(N = 3)。c)中的细胞外信号调节激酶(ERK)磷酸化1/2(P-ERK1 / 2)的由TNF-α和其增强的动力学的代表性免疫印迹和d)定量加入DMF(N = 3)。E)的c-Jun N-末端激酶的动力学的代表性免疫印迹(JNK)MAPK磷酸化(P-JNK)由TNF-α和在DMF的存在和不存在(N = 3)。F)DMF诱导HO-1的表达的代表性免疫印迹和其在TNF-α(N = 3)的存在下,通过将p38蛋白抑制剂SB203580减少。类似的结果在三个另外的细胞系,获得和图代表平均值±扫描电镜所有的实验。p值通过单因素方差分析计算,并用Bonferroni方法校正。
为了将DMF诱导的p38 MAPK磷酸化与HO-1表达联系起来,我们将DMF和p38 MAPK抑制剂SB203580或ERK1/2抑制剂PD98059一起处理asmc。抑制p38 MAPK可阻止DMF对HO-1的表达,而单独使用SB203580或PD98059对HO-1的表达无显著影响(图3 f).
DMF对p38 MAPK、HO-1和CXCL10的影响依赖于细胞内GSH
为了了解细胞内谷胱甘肽对DMF作用的作用,在添加任何其他处理之前,asmc先用谷胱甘肽- oet处理。GSH-OEt完全抑制DMF诱导的p38 MAPK磷酸化,并阻止DMF对TNF-α诱导的p38 MAPK磷酸化的加性作用,但对TNF-α单独作用无影响(图4).增加细胞内谷胱甘肽浓度可防止dmf诱导的HO-1在TNF-α缺失和存在时的表达(图4 b).所示图4 c, GSH-OEt处理显著降低DMF对TNF-α诱导的CXCL10分泌的抑制作用。
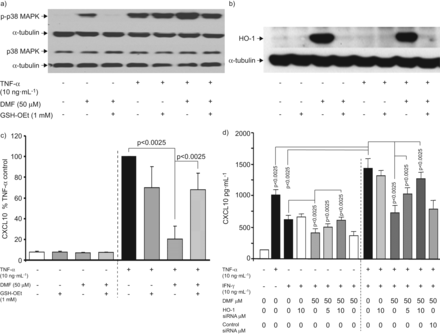
谷胱甘肽逆转二甲酯(DMF)的上p38丝裂原活化蛋白激酶(MAPK)磷酸化,血红素加氧酶的影响(HO)-1的表达和分泌CXCL10在气道平滑肌细胞(主动脉平滑肌细胞)。一个)的上DMF-谷胱甘肽乙基酯(GSH-OET)和肿瘤坏死因子的反转作用的代表性免疫印迹(N = 4)(TNF)-α诱导的p38蛋白的在主动脉平滑肌细胞的磷酸化刺激30分钟。B,C)GSH-OET对DMF依赖性HO-1的表达和TNF-α诱导的CXCL10分泌的抑制反转效果。数据表示为平均值±扫描电镜对4个独立细胞株进行3次重复实验,p值通过方差分析计算,并用Bonferroni方法进行校正。d) HO-1小干扰RNA (siRNA)可抵消DMF对TNF-α和IFN-γ诱导的CXCL10分泌的抑制作用。数据表示为平均值±扫描电镜对6个ASMC品系进行三次重复实验,用方差分析计算p值,并用Bonferroni方法进行校正。
DMF抑制TNF-α-和IFN-γ诱导的分泌CXCL10通过HO-1
INF-γ诱导CXCL10分泌,DMF抑制CXCL10分泌(图4 d).TNF-α和IFN-γ联合作用对CXCL10的分泌具有附加的刺激作用,DMF预孵育也可阻止该作用(图4 d).当用HO-1 siRNA预处理asmc时,DMF对TNF-α和IFN-γ诱导的CXCL10分泌的抑制作用完全被逆转(图4 d).对照siRNA (10 μM)没有改变TNF-α/IFN-γ诱导的CXCL10分泌或DMF的抑制作用(图4 d).
DMF剂量依赖性地抑制IFN-γ诱导的CXCL10分泌以及联合刺激的刺激效应(图5).补充谷胱甘肽可防止DMF在单独或联合IFN-γ刺激ASMCs中的抑制作用(图5).
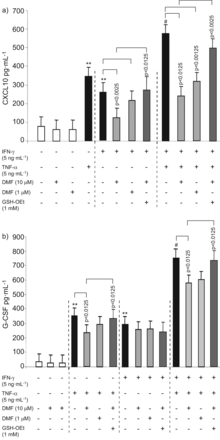
CXCL10和粒细胞集落刺激因子(G-CSF)通过二甲酯(DMF)的分泌和其反转由谷胱甘肽抑制是刺激 - 和细胞因子特异性的。一)六气道平滑肌细胞(ASMC)系用或者肿瘤坏死因子(TNF)-α或干扰素(IFN)-γ单独或两者结合处理。用DMF预处理剂量依赖性降低CXCL10分泌,其通过谷胱甘肽补充抵消。p值通过方差分析计算,并用Bonferroni方法进行修正。B)DMF对G-CSF分泌TNF-α和/或IFN-γ刺激在第24小时的效果和谷胱甘肽补充。数据表示为平均值±扫描电镜四种独立细胞系的三次重复实验。p值通过方差分析计算,并用Bonferroni方法进行修正。GSH-OEt:谷胱甘肽ethylester。**:与未受刺激ASMCs相比,p<0.01;#:P <0.01与单个刺激相比。
TNF-α和IFN-γ单独或联合诱导G-CSF分泌(图5 b).DMF单独对G-CSF分泌无影响,但部分减少TNF-α诱导的G-CSF分泌,而对IFN-γ依赖的G-CSF分泌无影响(图5 b).在TNF-α和IFN-γ刺激的asmc中,DMF治疗部分减少了G-CSF的分泌(图5 b).补充GSH-OEt可补偿DMF对TNF-α-的抑制作用,但对IFN-γ诱导的G-CSF分泌无抑制作用(图5 b).
讨论
在本研究中,我们提供的证据表明,DMF对培养的人原代asmc具有显著的免疫调节作用。DMF抑制TNF-α诱导的CXCL10,重要的是,当与氟替卡松联合使用时,显示了类固醇保留效应。DMF的抑制作用被介导通过诱导和p38蛋白,这随后诱导HO-1的表达的持续磷酸化。此外,IFN-γ诱导的分泌CXCL10,这是据报道,在主动脉平滑肌细胞对类固醇不敏感,也通过DMF抑制。提高GSH DMF之前治疗的细胞内水平反转所有药物的效果。因此,我们的数据表明,DMF有潜力用于治疗慢性炎症性肺部疾病的治疗。
哮喘气道中ASMC数量和大小的病理增加是由H超级和K.essl发表[15.这一发现后来被证实出现在儿童哮喘中,通常在发现任何炎症迹象之前[16.,17.].在哮喘中,ASMCs在暴露于各种各样的诱因下会收缩和变窄气道,从而限制气流。此外,ASMCs通过分泌大量促炎细胞因子促进局部炎症[3.].基于这些观察,靶向ASMC活性和细胞因子分泌可能有助于解决慢性肺部疾病的炎症。
我们观察到DMF下调ASMCs CXCL10的分泌尤为重要,因为CXCL10是肥大细胞进入炎症气道组织的最强招募因子之一[4- - - - - -6].在这项研究中,我们发现,DMF抑制IFN-γ诱导CXCL10通过HO-1的表达。这一观察结果加强了在哮喘治疗中的治疗用途DMF作为IFN-γ诱导的分泌CXCL10的论点是不敏感的类固醇治疗[9].此外,在TNF-α激活的人气管平滑肌的DMF与氟替卡松的组合的还原效果比单独使用每种药物的更强。
人肺成纤维细胞和主动脉平滑肌细胞,我们已经表明,抑制DMF PDGF-BB和TNF-α诱导的IL-6 [分泌18.,19.].这一效应是由成纤维细胞中DMF抑制核因子-κB p65和激活蛋白-1两种转录因子介导的[18.,19.].我们进一步提供了DMF降低组蛋白3和cAMP反应元件结合蛋白(CREB)磷酸化的证据[18.,19.].DMF对NF-κB和CREB的抑制作用可以解释其对IFN-γ诱导的分泌CXCL10效果,因为这两个转录因子介导IFN-γ信号传导到CXCL10活性9,20.].这些发现表明了DMF通过减少几个刺激基因表达的转录因子的结合的更一般的作用模式。
此前,我们报道DMF通过诱导HO-1显著降低pdgf - bb诱导的asmc增殖,提示DMF可能降低气道重塑[13.].在本研究中,我们感兴趣的是DMF介导的HO-1上调是否也参与了DMF的抗炎作用。HO-1在其他过敏性疾病中具有有益的抗炎作用,并可能表明DMF具有全面的保护性抗炎作用[21.,22.].与我们的结果类似,吡咯烷二硫代氨基甲酸盐诱导HO-1表达,从而降低支气管肺泡灌洗液中的嗜酸性粒细胞和t细胞计数,以及过敏性气道疾病小鼠模型中的气道高反应性[23.].所观察到的嗜酸性粒细胞减少的活性可能与我们的发现,即DMF通过阻断组蛋白3磷酸[减少趋化因子RANTES和的分泌19.].增加HO-1在炎症肺中的表达的进一步有益作用可能与减少气道炎症、粘液分泌、氧化应激和气道超反应有关,这些在卵蛋白致敏的豚鼠哮喘模型中得到了证实[22.].在我们的研究中,DMF在TNF-α存在的情况下诱导HO-1,而在HO-1 siRNA处理过的asmc中,这种作用被取消。此外,另外两种HO-1诱导物,haemin和钴原卟啉,也能减少TNF-α诱导的CXCL10分泌,从而支持HO-1介导DMF的抗炎作用的假设。
根据我们之前的研究,我们发现DMF通过p38 MAPK磷酸化增强TNF-α诱导的HO-1表达。此外,补充谷胱甘肽补偿了DMF对p38 MAPK磷酸化和CXCL10分泌的影响。我们的发现与另一项研究一致,即谷胱甘肽消耗增加了C6胶质瘤细胞中p38 MAPK的磷酸化[24.].此外,DMF降低GSH水平,从而抑制外周血单个核细胞分泌IL-12和IFN-γ [10.].在人肺成纤维细胞中,细胞内谷胱甘肽的减少诱导HO-1 [25.].此外,在胰腺星状细胞,p38蛋白的活化增强HO-1的表达[26.].在CXCL10分泌方面,HO-1降低了分离的巨噬细胞CXCL10的表达[27.].
为了进一步表征DMF的信号特异性作用,我们使用IFN-γ (CXCL10的一种已知诱导物)刺激细胞[9].我们证实,IFN-γ与TNF-α结合对CXCL10分泌和G-CSF分泌的添加剂刺激作用[9,28.].这些研究表明,NF-κB激活介导TNF-α诱导的CXCL10分泌,但仅在IFN-γ刺激细胞时略微参与。我们的数据表明,DMF通过降低细胞内谷胱甘肽水平控制人ASMCs TNF-α和IFN-γ诱导的CXCL10分泌。
总之,我们在这里描述了一种新的分子信号机制,通过DMF抑制asmc细胞因子诱导的CXCL10分泌。此外,我们还发现DMF具有节省类固醇的特性。因此,DMF可以被认为是慢性炎症性气道疾病免疫反应的调节剂。
致谢
我们感谢C.T. S'ng(巴塞尔大学医院,巴塞尔,瑞士)他的帮助准备这份手稿。
脚注
支持声明
这项研究得到了瑞士国家基金会的不受限制的研究资助(批准310030 - 130740/1),以及瑞士巴塞尔Gottfried和Julia Bangerter Ryhner-Stiftung在2010年给M. Roth的哮喘研究资助。
感兴趣的语句
没有宣布。
- 已收到2011年4月21日。
- 接受2012年4月4日。
- ©2013人队











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}