





摘要
肺部的急性炎症对宿主防御至关重要,但慢性或过度炎症会导致几种常见的呼吸系统疾病,包括哮喘和急性呼吸窘迫综合征。
炎症的消退是一个积极的过程。在健康方面,急性炎症发作时的事件为特定的化学介质建立了生物合成回路,这些介质后来充当激动剂,以协调组织内稳态的回归。除了过度的促炎刺激,病理性炎症也可以由分辨率信号的缺陷引起。
对抗炎、促溶解分子及其反调控信号通路的理解为肺部疾病的分子病理生理学提供了新的见解,并为治疗策略的设计提供了机会。
在本综述中,研究了不断增长的脂质溶解介质家族,包括脂素,resolvins, protectin,环戊酮和前qualene二磷酸。这些化合物或其结构类似物在调节气道炎症中的作用已被发现。
肺部和气道炎症在欧洲和美国都是一个重大的医疗和经济负担1.哮喘是炎症性肺病中发病率最高的疾病,自20世纪80年代以来发病率翻了一番2.哮喘的特征是气道内嗜酸性粒细胞、t细胞和肥大细胞浸润,粘液过多,在某些情况下,气道重塑与平滑肌改变,这些共同导致气流阻塞的临床特征。急性呼吸窘迫综合征(ARDS)是死亡率最高的炎症性肺部疾病3..ARDS的特征是广泛的炎症和多形核白细胞(PMN)在肺部的激活3..哮喘的慢性炎症和急性呼吸窘迫综合征的强烈急性炎症反应代表了炎症性肺部疾病谱系的两个不同的端点,然而这两种呼吸系统疾病的特征都是无法限制炎症。
炎症是对组织损伤、感染和过敏原挑战的一种生理反应,其进化目的是限制有害物质的损害和传染性生物的传播4.炎症反应具有内在的保护作用,密切参与组织内稳态的恢复。直到最近,炎症的解决一直是炎症研究的一个代表性不足的焦点。现在很清楚的是,炎症反应的缓解阶段是一个积极的和精心安排的过程,在复杂性上类似于炎症的发生和维持5.在恢复组织稳态时,化解不仅能抑制炎症,还能促进免疫防御机制。溶解的过程起源于炎症反应的开始,随着生物合成回路的建立,随后产生反调节化学介质。促进溶解的分子本质上不同于单纯的抗炎化合物,因为促溶解分子有助于组织降解,使组织恢复正常6,7.
在本研究中回顾了五类自然发生的前分解分子(图1)⇓),除了它们的信号通路,在恢复组织稳态中的作用和对肺部炎症的细胞效应因子的影响(表1⇓).
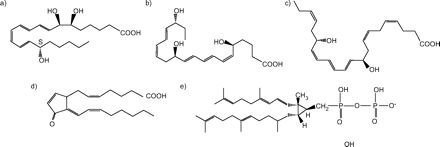
内源性抗炎脂类。内源性抗炎脂质的五个类别的代表成员显示:a)脂素,脂素a4;b) resolvins, resolvin E1;c) protectins, protectin D1;D)环戊烯酮,15deoxyΔ12 - 14前列腺素J2;e)聚异戊二烯磷酸,二磷酸前夸烯。
分解的化学介质
Lipoxins
脂素(LXs)是花生四烯酸(C20:4)代谢过程中脂氧合酶(LO)相互作用的产物,其结构和生物学性质不同于其他类二十烷酸35,36.LXs具有强大的抗炎特性,对白细胞、内皮细胞、上皮细胞和其他基质细胞具有细胞类型特异性作用(图2)⇓;表1⇑).在哮喘和ARDS中,LXs抑制嗜酸性粒细胞的转运,这与缓解炎症特别相关12,13,37PMN的趋化性、毛细后小静脉的反迁移、超氧阴离子的产生以及亲azurofi颗粒的脱粒6.此外,LXs刺激巨噬细胞清除凋亡的pmn18,阻断自然杀伤细胞的细胞毒性和肿瘤坏死因子(TNF -α)从t细胞释放14,15.
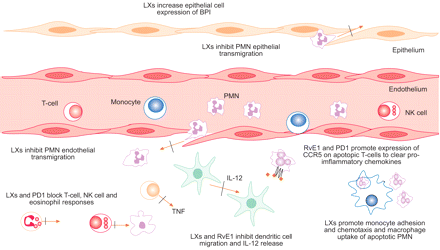
一些脂质介质的细胞型特异性反调节作用。内源性类autacoids显示细胞类型特异性的作用,以促进炎症。脂素(LXs)与特定受体接合(如。ALX)可抑制多形核白细胞(PMN)通过内皮细胞和上皮细胞的转运,并可抑制先天免疫效应物的促炎反应,包括PMN、t细胞、嗜酸性粒细胞和自然杀伤细胞(NK细胞)。LXs也增强了凋亡PMN的清除。值得关注的是,这些化合物增加了粘膜上皮细胞杀菌/通透性增加蛋白(BPI)的表达,以抵御病原体。resolvin (如。resolvin E (RvE)1)和protectins (如。保护素D (PD)1也显示细胞类型特异性的反调控反应,以促进解决。CCR: CC趋化因子受体;TNF:肿瘤坏死因子;IL:白介素。
lx形成了通过中间体在细胞间双向转移的跨细胞生物合成38.可以生成LXs通过至少有三条不同的路径。一种途径涉及白细胞5- lo催化C20:4转化为白三烯(LT)A4,在脉管系统中,随后被血小板吸收并转化为LXA4由12-LO39.第二种途径包括上皮细胞、嗜酸性粒细胞或单核细胞来源的C20:4通过15- lo转化,产生15(S)-氢过氧二十碳烯酸,它也可以作为白细胞5-LO的底物。该反应生成一种不稳定的环氧四烯中间体,可通过水解酶转化为LXs35,40.5-LO衍生LTA4也可以通过15-LO转换为LXs。虽然这三条途径是LX生成的主要途径,但可能还存在其他5- lo独立的途径。
有趣的是,阿司匹林,主要的非甾体抗炎药,抑制前列腺素(PG)的合成,但剂量远低于发挥其抗炎作用所需的剂量41.最近,阿司匹林引发的15-epimer-LXs (ATLs)的发现解决了这一悖论。42.阿司匹林乙酰化环加氧酶(COX)-2的活性位点以抑制pg的产生,但该酶仍然能够将C20:4转化为15(R)-羟基二十氧乙酸(15R-HETE)。该化合物可作为5-LO的底物进一步转化为ATLs42.15-epimer-LXs增加一氧化氮的合成通过组成性或炎症性一氧化氮合酶,一氧化氮减少白细胞-内皮细胞相互作用抑制炎症组织内白细胞积累43.因此,阿司匹林可以通过抑制促炎PG的生物合成和促进抗炎15-epimer-LXs的形成来发挥抗炎作用。在没有阿司匹林的情况下,细胞色素p450酶也可以产生15R-HETE,作为15-epimer-LX跨细胞生物合成的底物44,45.
LXs在15-羟前列腺素脱氢酶和PG还原酶的作用下代谢失活,形成13,14-二氢-15-羟基- lxa49,46.LX和15-epimer-LX代谢具有立体特异性,其中15-epimer-LXs代谢效率较低,因此这些ATLs的生物半衰期大约增加了两倍46.已经产生了抗失活的LX类似物9.这些修饰增强了LXs的生物活性,已被证明是研究LXs生物学功能的有用工具在体外而且在活的有机体内.
LXs不仅发挥着指导白细胞功能向分辨率的作用,而且还可以向局部基质微环境的分辨率发出信号(表1)⇑).正常人支气管上皮细胞(NHBE)暴露于盐酸后,COX-2和高亲和力LXA的表达增加4受体ALX。LXA4通过增加基础NHBE增殖和抑制分化NHBE中的促炎事件,如细胞因子释放和PMN转运,促进酸损伤的恢复26.尽管LX可有效调节上皮细胞和白细胞功能,但其生物作用不同于免疫抑制化合物,因为LX信号通路可调节病原体介导的炎症20.,47促进粘膜细菌杀灭通过细菌/渗透诱导蛋白(BPI)在上皮细胞中的表达22.因此,除了抗炎,LXs还具有宿主保护作用。
LXA4受体
LXs与一个或多个特定受体相互作用,包括它们自己的特定受体,LTD的一个子类4受体(即。半胱氨酸(Cys)LT1),以及其他细胞内识别位点48,49.的LXA4受体ALX是一种结合LXA的g蛋白偶联蛋白4具有较高的亲和力(KD= 1.7 nM)48.ALX是最初被鉴定为结合脂质和肽配体的受体48,50.在pmn中,ALX信号传导部分发生,通过聚异戊二酯磷酸(PIPP)重塑(见下页)10抑制白细胞特异性蛋白-1磷酸化,这是p38-丝裂原激活蛋白激酶级联的下游调节因子(表2⇓)51.
ALX介导膜联蛋白-1抗炎信号
糖皮质激素是有效的抗炎分子,通过抑制促炎介质的产生在解决炎症中发挥主要作用58,59减少白细胞粘附分子的表达60.地塞米松还能促进单核细胞和t细胞从发炎组织中迁移61,62.糖皮质激素作用通过在细胞质内的同源受体,当配体结合时,移动到细胞核以调节转录63.在哮喘治疗中,皮质类固醇可诱导ALX的表达64以及annexin-1,它也可以与ALX相互作用以启动抗炎信号65.
膜联素-1是一种有效的抗炎分子,在pmn中大量表达66.大部分的膜联蛋白-1在细胞质内。当PMN激活并粘附在发炎的血管内皮上时,膜联蛋白-1迅速外化67导致细胞脱离发炎的血管68减少PMN的招募。在急性炎症模型中,向annexin-1添加抗血清可导致pmn在炎症渗出液中持续存在69.在annexin-1敲除小鼠中,pmn更容易被激活28,69.一些annexin-1及其n端肽Ac2-26的抗炎作用是由与ALX的直接相互作用介导的,但具有较低的亲和力(KD= 900 nM)470.
LXs在肺部疾病中的作用
LXs是在一系列呼吸系统疾病期间在肺部产生的71.有趣的是,LXs的低生物合成能力与严重的气道炎症有关。阿司匹林加重的呼吸系统疾病是一种更严重和持久的哮喘形式,在全血中,与阿司匹林耐受哮喘患者相比,这些哮喘患者产生LXs的能力下降72.此外,低水平的LXA4重度哮喘患者诱导痰上清液中是否存在轻度哮喘患者73.此外,严重哮喘患者在全血中将C20:4转化为15- lo催化产物(包括15-HETE和LXA)的能力降低474.与15-LO活性下降形成鲜明对比的是5- lo衍生产物,包括5-HETE, LTB4和cyslt,在严重哮喘中都增加了74.1秒用力呼气量百分数预测值及循环LXA水平4与cyslt相关,提示这些生物活性脂质介质的生物合成能力与哮喘气流阻塞之间存在联系。因此,在严重哮喘中,LX生成的减少和LT生成的增加造成了一种不平衡,维持了这种情况下典型的持续性气道炎症和气流阻塞。
在一些患有囊性纤维化的患者的气道中也发现了LXs水平的降低75.因为在慢性支气管炎中也得到了类似的结果,气道中LX形成的改变可能代表了慢性pmn富集的气道炎症的更广泛的后果76.为了支持LXs可能在治疗呼吸道炎症方面提供可行的治疗策略的概念,用稳定的LXA治疗小鼠4在过敏性气道炎症和急性肺损伤(ALI)小鼠模型中,与CD11b启动子组件偶联的人类ALX转基因小鼠减少了白细胞浸润。37,77.
pg和lt
与LXs一样,pg和lt也从C20:4中酶促衍生,并作为有效的脂质介质78.它们在正常生理中起着至关重要的作用,并在急性炎症的早期发挥重要作用。在炎症发作时,C20:4由cox代谢为PGs,如PGD2,铂族元素2, PGF2α, PGI2血栓素A2众所周知,它们对炎症细胞和肺组织具有强大的生物学作用。除了已被充分描述的pg在促进炎症反应方面的作用外,最近的研究还强调了cox -2衍生pg在缓解炎症方面的抗炎和抗纤维化作用77,79,80.在急性缓解胸膜炎症的模型中,炎症发作后COX-2水平在早期(2小时)和晚期(48小时)短暂升高,以产生COX-2衍生的PGD2和15脱氧Δ12 - 14PGJ2(15 d-pgj2)79.COX-2-derived铂族元素2还能解决过敏性胸膜炎吗79,80以及分泌物中tnf启动的PMN激活81.有趣的是,COX-2衍生的PGD2和铂族元素2能诱导15-LO表达促进LX生物合成吗81.在自发解决ALI的模型中,选择性COX-2抑制或缺乏导致炎症延长,部分原因是通过减少PGE的产生2以及前化解介质,包括LXA4和15-epimer-LXA477,79.除了pg外,lt还具有对白细胞和呼吸组织重要的促炎特性82.5-LO对C20:4的代谢导致了lt的形成。英国网球协会4是一种不稳定但关键的中间体,可以转化为LTB4由本公司4水解酶,LTC4LTC的4合成酶或通过12-或15-LO到LXs6.特别是LTB4是一种有效的PMN趋化剂和促分泌剂,而LTC4和有限公司4强效支气管收缩剂82.有趣的是,LTB4对宿主防御、促进PMN吞噬致病菌和抗病毒机制是否重要83,84.cysltts不具有这些相同的特性,因为小鼠缺乏多药耐药蛋白1,该蛋白与LTC的细胞挤压有关4,对链球菌引起的肺炎全身的肺炎85cyslt1受体选择性拮抗剂在脓毒症动物模型中提供了生存优势86.
环戊烯酮
cox衍生中间体PGG2可以通过PGD转换2合成酶转化为PGD2它对白细胞和气道组织表现出促炎和刺激作用87.然而,PGD2进一步脱水生产J系列的pg,包括Δ12 - 14-PGJ2(PGJ2)和15d-PGJ2.有人提出,在炎症消退过程中诱导COX-2对环戊酮pg (cyPGs)的形成是必要的,后者可以介导反调节作用通过过氧化物酶体增殖物激活受体(PPAR)-γ的激活(表2)⇑)54,79.比如环戊烯酮15d-PGJ2抑制血管细胞粘附分子(CD106)和细胞间粘附分子(CD54)在人脐静脉内皮细胞上的表达88.15d-PGJ的作用2与LXs相比,它们抑制而不是增强巨噬细胞的活性,而对pmn几乎没有影响(表1)⇑)89.此外,15d-PGJ的药理学浓度2能抑制淋巴细胞增殖和白细胞介素-2的产生吗90,91.这些细胞类型特异性的作用被15d-PGJ2阻止单核细胞粘附到人主动脉血管,而对PMN没有影响。15 d-pgj2也抑制CC趋化因子配体(CCL)2(单核细胞趋化蛋白-1)在内皮细胞上的表达,但不抑制CCL8 (IL-8)27.
resolvin
Resolvins(溶解相相互作用产品)是欧米茄3脂肪酸衍生的抗炎脂质,最初是在自发溶解的渗出物中发现的95,96.Resolvins根据脂质来源分为不同的系列。D系列的Resolvins (如。RvD1)来源于二十二碳六烯酸(DHA;C22:6)和E系列的resolvins (如。RvE1)来源于二十碳五烯酸(EPA;C20:5)6.
Resolvins在人全血中通过酶促DHA转化为含17s -羟基的d系列Resolvins生成。在阿司匹林的存在下,这些化合物的生成显著增加95,96.内皮细胞在低氧条件下生长,用阿司匹林处理,通过阿司匹林乙酰化COX-2将DHA转化为17r -羟基DHA,产生17R-Resolvin D系列。17r -羟基- dha也是人类PMN的底物,可以形成两组二羟基和三羟基产物。17r -羟基- dha在碳7处的酶氧化作用导致阿司匹林触发的RvD1 (at -RvD1)和at - rvd2。当17r -羟基- dha与碳4氧化时,生成at - rvd3和at - rvd46.在体外经阿司匹林处理的内皮细胞和脑源性微胶质细胞将EPA转化为18r -羟基二十碳五烯酸(18R-HEPE)和15R-HEPE。18R-和15R-HEPE都可以被活化的PMN迅速转化为含有5(6)-环氧化合物的分子,然后转化为具有生物活性的5,12,18r -三羟基二十碳五烯酸(RvE1)。95.
D系列和E系列的Resolvins都具有有效的抗炎特性,如抑制PMN迁移和缩短急性炎症的消退期97(表1⇑).第一个RvE1受体被确定为ChemR2331.ChemR23在单核细胞、巨噬细胞和树突状细胞(dc)上表达。用RvE1治疗可抑制炎性结肠炎、DC迁移和IL-12产生,并减弱核因子-κB激活(表2)⇑)30.,31.有趣的是,ChemR23最初被发现是一种趋化蛋白的受体,这种趋化蛋白存在于炎症分泌物中,称为趋化素98.最近,发现了RvE1的第二种受体,即LTB4受体BLT1,在pmn、嗜酸性粒细胞、单核细胞和t细胞上表达99,One hundred..
令人感兴趣的是,老鼠已经被开发出了ω -3脂肪酸去饱和酶的转基因,从而从ω -6脂肪酸中产生更多的ω -3脂肪酸101.这些转基因动物在胃肠炎症模型中免受结肠炎的侵害102.这种保护作用不是继发于促炎脂质PGE的减少2和LTB4.相反,resolvins的水平显著增加102.Omega-3脂肪酸集中在鱼油中,富含Omega-3脂肪酸的饮食可以预防哮喘、囊性纤维化、心脏病和癌症103- - - - - -105.这些新发现的omega-3化解素为这些有益的作用提供了潜在的分子原理。
神经保护素D1/保护素D1
Protectin (P)D1是由DHA在15- lo催化反应中生成的10R, 17s -多沙二烯108,109.在急性、自发消退的小鼠腹膜炎中,已经定义了新的消退指数,包括ψ马克斯,(存在pmn的最大数量),t马克斯(当ψ的时候马克斯发生)和R我,(分辨率区间为t马克斯来t50),使PMN数达到ψ的一半马克斯)97.使用这种方法,PD1在分辨率渗出液中增加,R有最有效的降低我与LXs相比,15-epimer-LXs和e系列分辨率较高97.这种二十二烯后来被命名为神经保护素D1/保护素D1,在大脑中发现了大量DHA的神经胶质细胞,并对缺血性脑损伤和阿尔茨海默病具有保护作用108,110,111.PD1还可以调节巨噬细胞和结构细胞的反应,以保护肾和肝损伤(表1)⇑)29,112.
在肺部疾病中的作用
在哮喘加重期间,PD1及其生物合成中间体17s -羟基- dha存在于呼出的冷凝物中,但与健康受试者的呼出冷凝物相比,其含量显著降低113.此外,PD1也存在于炎症的小鼠肺匀浆中。当在气溶胶过敏原挑战之前给过敏原致敏小鼠注射外源性PD1时,PD1显著阻断白细胞浸润和气道高反应性。当过敏性气道炎症已经建立后,PD1也可以加速嗜酸性粒细胞的清除。因此,PD1具有有趣的特性,为治疗哮喘提供了令人兴奋的新治疗策略。
Polyisoprenyl磷酸盐
许多呼吸道的炎症性疾病都与大量的pmn有关,包括ARDS、肺炎和严重哮喘3.,114,115.组织浸润性pmn可对周围组织造成广泛损害,并使炎症持续存在通过有害物质的无意释放,如超氧阴离子和蛋白酶4.重要的是,存在控制PMN激活的自然机制。PIPPs作为PMN的“停止”信号起着独特的作用。PIPPs存在于PMN膜中。pmn的激活启动PIPP重构,并迅速转化为其单磷酸形式的前qualene二磷酸(PSDP) (PSMP)34.PSDP,而不是PSMP,显著减少PMN释放的超氧阴离子生成(表1⇑)10,34.
已经发现了PIPPs的几个调控位点。PSDP抑制植物、微生物和哺乳动物磷脂酶(PL)D(表2)⇑)34,116.这种酶将磷脂酰胆碱转化为磷脂酸。117它在PMN中携带重要的细胞内信号,导致广泛的功能反应,包括肌动蛋白重塑、颗粒释放和烟酰胺腺嘌呤二核苷酸磷酸氧化酶的激活。磷脂酰肌醇3-激酶(PI3K)活性对早期细胞活化很重要118, PSDP最近被确定为PI3K的调节因子57.LTB4刺激PMN PSDP水平的快速下降和PI3K活性的增加。PSDP与p110γ-PI3K相互作用,使其失活在体外,PSDP对重组p110γ-PI3K表现出浓度依赖性抑制。PSDP结构模拟物可以抑制PLD, PI3K, PMN反应和肺部炎症在活的有机体内57,116.
PSMP不具有PSDP的强大抑制特性。PMN激活后,PSDP通过含有2的PA磷酸酶结构域转化为PSMP119.这种酶的功能特征是磷酸酶,它将PSDP和法尼二磷酸转化为它们的单磷酸,因此最近被重新命名为聚异戊二磷酸磷酸酶(PDP)-1(图3)⇓).PDP-1在pmn和许多人体组织中表达,是脂质磷酸酶/磷酸转移酶(LPT)家族的成员120.LPTs有5个亚群,包括脂质磷酸磷酸酶、sphingomyelin -1-phosphate (S1P)磷酸酶、sphingomyelin synthases、脂质磷酸酶相关蛋白/可塑性相关基因和一个没有任何预先指定功能的亚群,称为候选sphingomyelin synthases type 2 (CSS2)。PDP-1是lpt的CSS2家族的成员119.
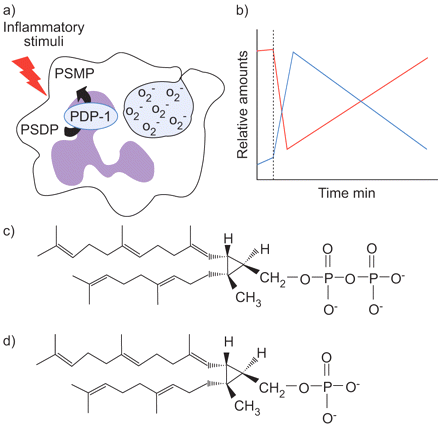
a)说明和b)前qualene二磷酸(PSDP)重塑人类多形核白细胞(PMNs)的时间过程。PSDP是一种细胞内的反调节分子,以纳摩尔的量存在于新分离的pmn中。炎症刺激的激活(烧嘴句子)导致PSDP(红色)迅速而短暂地转化为单磷酸前qualene (PSMP;蓝色)与功能反应同时发生,如超氧阴离子(O2-)的一代。PSDP重构是由PDP-1 (PDP-1)介导的。随着细胞失活,PSDP池在几分钟内恢复。c) PSDP的化学结构;d) PSMP的化学结构。
除pmn外,PDP-1在主要免疫器官(肺、脾、胸腺、肝、肠)中也有高表达。这表明PDP-1不仅在髓系细胞中发挥重要作用,而且在结构细胞、巨噬细胞、dc和淋巴细胞中也发挥重要作用。最近研究表明,S1P在控制淋巴细胞从胸腺、淋巴结和脾脏排出方面起着至关重要的作用121.S1P由S1P磷酸酶代谢,这表明LPTs可能在控制免疫反应中发挥许多重要作用。
PSDP在肺部疾病中的应用
在吸酸引起的肺损伤和炎症的实验模型中,存在相互关系在活的有机体内对肺PSDP和PI3K活性的影响57.开发了一种新的二膦酸盐PSDP结构模拟物来抵抗磷酸酶为基础的失活,这种PSDP模拟物阻断了LTB对人体PMN的激活4以及小鼠肺PI3K活性和炎症。这些发现表明PSDP是一种内源性PI3K抑制剂,并提示在以PMN过度激活为特征的炎症性疾病中,PIPPs可以作为抗PMN治疗策略的结构模板,以限制与ARDS相关的组织损伤。
分辨率中的细胞事件
细胞凋亡和吞噬作用
在炎症反应期间,在组织中积累的免疫细胞数量大幅增加。除了阻止进一步的白细胞招募,已经存在的白细胞可以退出炎症组织通过引流淋巴或进行程序性细胞死亡,由吞噬细胞清除非炎122.凋亡与炎症的缓解有着内在的联系,凋亡的损害或凋亡细胞的清除都会导致慢性炎症和自身免疫123.类似地,细胞凋亡的增强可以加速炎症反应的消退124.LXs促进巨噬细胞吞噬凋亡的pmn18.这一过程通过释放抗炎转化生长因子-β进一步调节炎症环境,可以将naïve前体转化为调节性t细胞125.吞噬凋亡细胞后,巨噬细胞也会释放反调节脂质介质,包括PGE2, PGF1-α和LXs126,127.
分解过程中白细胞的排出
在炎症反应开始时,细胞被特定的趋化剂招募到炎症焦点,这些趋化剂协调白细胞亚群的招募,从pmn开始,然后是嗜酸性粒细胞、单核/巨噬细胞和淋巴细胞128,129.调节趋化因子的产生对于白细胞的正常生理迁移和炎症反应期间白细胞的募集至关重要130.改变趋化因子的产生通常是慢性炎症的一个特征,在受累组织中白细胞的募集和保留增加131.炎症微环境通过特定趋化因子受体的表达调节白细胞的排出,如。t细胞可以利用CC趋化因子受体7退出通过传入淋巴管132,133.同样,通过增加趋化因子受体CXCR4的表达,PMN可以以基质来源因子-1导向的方式从炎症组织中清除,并返回骨髓134.前分解脂质介质是趋化因子表达的有效调节因子33.用选择的趋化因子修饰组织间质可以保留特定的白细胞并降低分辨率135,136.
当炎症消退时,需要从微环境中去除趋化因子以阻止白细胞进一步招募。为此,“沉默”趋化因子受体D6在清除炎症,但不是组成型趋化因子中发挥重要作用137.D6敲除小鼠不能解决急性炎症反应,这表明D6在解决炎症反应中具有重要作用138.浸润的白细胞也可以上调选择的趋化因子受体,因为它们正在经历凋亡,以隔离趋化因子,从而防止进一步的白细胞募集33.LXs是增加凋亡pmn清除的有效刺激,但关于LXs或促溶解脂质对白细胞排出的影响知之甚少18.
炎症和宿主防御的解决
炎症的解决不同于免疫抑制,部分是通过促进宿主防御。有趣的是,LXs通过控制病原体诱导的炎症反应来增强粘膜宿主防御20.,47诱导BPI的表达22.BPI是一种由PMN和上皮黏膜释放的55 kDa蛋白。BPI破坏革兰氏阴性菌的内外脂膜,增强细菌的吞噬作用,隔离脂多糖139.上皮细胞暴露于LXA4或ATL上调BPI,增加胃肠上皮对沙门氏菌的杀伤22.LXA4和RvE1也能保护兔子免受Porphyromonas gingivalis-诱导牙周病,表明前化解分子增强,而不是损害,宿主防御在活的有机体内140.
治疗的影响
目前许多炎症性疾病的治疗靶点集中在阻断炎症的起始或放大介质。虽然这一策略在某些临床情况下是有益的,但对于常见的炎症性肺部疾病,包括哮喘和急性呼吸窘迫综合征,仍有大量的临床需求未得到满足。替代治疗策略可能不是早期阻断或选择促炎介质,而是强调模拟LXs、cyPGs、resolvins、protectins、PSDP或其他加速炎症消退的天然反调节分子。其中一些化合物的代谢稳定类似物已被开发出来并显示出效力在活的有机体内几种模型系统中的保护作用116,141.
有趣的是,抗炎疗法可以影响促溶解化合物的形成或作用。例如,阿司匹林促进15-epimer-LXs和resolvins的形成17,96.由于担心诱发哮喘,这种药物很少使用,尽管它有潜在的治疗效果142.糖皮质激素通常用于控制哮喘,除了增加ALX配体annexin-1 (维达在上),可增加ALX受体的PMN表达64.此外,一些抗炎实验药物,包括5- lo相互作用蛋白(FLAP)抑制剂(BayX-1005),既可以降低LT,又可以增加LX的形成在活的有机体内143.
结论
炎症的消退是对组织损伤和感染的生理反应的一个组成部分。解析机制的阐明导致了其作为一个基本的稳态过程的认识。在健康方面,在急性炎症反应刚开始时就开始建立化解信号通路。促分解介质形成的动力学是高度调控的,并与炎症中的细胞运输事件有关。内源性化学激动剂的发现及其信号通路为开发新的治疗策略和进一步了解慢性和严重炎症性肺部疾病(如哮喘和急性呼吸窘迫综合征)的病理生理学提供了机会。更好地理解气道炎症的解决机制可能为与这些和其他常见呼吸道疾病相关的过度发病率和死亡率提供新的治疗方案。
致谢
作者要感谢C. Schneider (Brigham and Women 's Hospital, Boston, MA, USA)在手稿准备方面的专家帮助。
- 收到了2007年1月16日。
- 接受2007年6月15日。
- ©ERS期刊有限公司











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}