





抽象的
非常长链酰基-CoA脱氢酶缺乏(VLCAD)是催化线粒体β-氧化中长链脂肪酸的脱氢的关键酶。VLCAD缺乏是一种遗传症,其常见的婴儿期或儿童患者,具有噬菌体低血糖,心肌病和肝功能障碍的发作。
本研究报告了一名18年代的中国女性,在一段时间内呈现急性高潮呼吸衰竭和横纹肌溶解的速度。培养的成纤维细胞的VLCAD活性降低了VLCAD缺乏症。
患者完全恢复了支持性护理。急性发作后肺功能试验显示慢性亚临床呼吸肌弱点的证据。
总之,在患有无法解释的急性呼吸麻痹和失败的患者中应考虑这种罕见的代谢障碍。
代谢肌病包括一组异质遗传紊乱,导致肌肉组织中的能量产生缺陷。其中是脂肪酸氧化和糖原储存疾病的障碍。非常长链酰基-CoA脱氢酶(VLCAD)缺乏是一种稀有的遗传酶疾病,其中线粒体的非常长链脂肪酸的β-氧化中的第一和速率限制步骤有缺陷(图1⇓)。vlcad缺乏有三种表型,取决于年龄和介绍,这可能发生:1)在新生儿用低钾性低血糖,肝功能障碍和心肌病;2)后期在婴儿期或儿童时期与低钾低血糖发作和肝功能障碍;3)在患有运动不耐受的青少年或成人中,复发性肌球蛋白血症和横纹肌分解(表1⇓)。
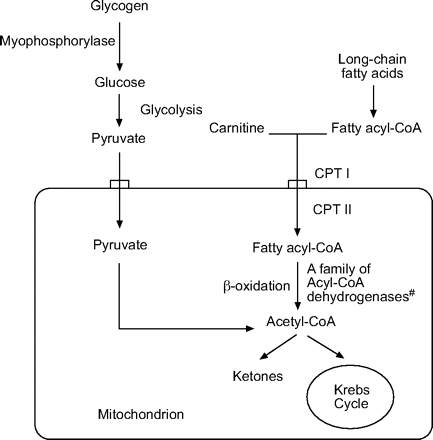
脂肪酸β-氧化途径。CPT:肉毒氨酸棕榈酰丙烷酶。#:β-氧化的反应通过针对不同链长的酰基-CoA脱氢酶催化,包括非常长链,长链,中链和短链酰基-CoA脱氢酶。
在这里,本作者报告了横纹肌溶解和急性肌肉瘫痪的案例,导致中国女性呼吸衰竭,以前未确诊的VLCAD缺乏。
案例报告
一个明显健康的18岁女性被允许持续1天的持续肌肉疼痛和弱点。入场前的一天,她在16:00吃晚饭,从那时起没有吃过。第二天早上,患者醒来伴随着全天的广义肌肉疼痛和弱点。
那天晚上,她终于带到医院。当她入院时,她没有吃过> 24小时。
患者患有类似的肌肉疼痛和弱眠发作的历史,并且自从10年龄龄的衰弱,却经暗尿液,但严重程度较小。这些通常由中等运动带来。她否认了任何草药或毒品的摄入,没有类似条件的家族史。在这种情况下,肌痛和弱点比以前的发作更严重。
在考试时,患者完全有意识但出现弱。她随着126/77 mmhg的血压,脉冲110击球·min-1,呼吸速率16呼吸·分钟-1,室内空气中的动脉氧饱和96%。神经学检查揭示了4/5级的广义肌肉弱点,反射减少,四肢均为低渗。呼吸系统和其他系统的考试是不起眼的。
胸部射线照相显示正常的心脏阴影和透明肺部。实验室调查显示:白细胞计数24.5×109.·L.-1;钠133mmol·L.-1;钾5.8mmol·L.-1;肌酸激酶224,500 u·l-1;乳酸脱氢酶10,030 u·l-1;和丙氨酸转移酶909 u·l-1。尿液分析对酮负阴性。
在入院时间内,随着渐进式呼吸麻痹和失败的发展,她的肌肉弱点恶化。动脉血气测量,而患者在补充氧气上显示出以下:呼吸酸中毒和Hypercapnia的存在;pH 7.28;动脉氧张力/吸气氧比比56.76kPa;二氧化碳动脉张力15.84 kPa;基础超出-4.5mmol·L-1;和碳酸氢盐22.7mmol·L-1。她需要气管内插管和正压力通风。在重症监护病房中,血清尿素和肌酐在6.1mmol·L达到峰值-1和118μmol·L.-1, 分别。患者的病情稳定,并在10天后成功地从呼吸机断奶。
怀疑通过禁食和锻炼沉淀出横纹肌分解代谢肌病的诊断。进一步调查显示:血清乳酸1.2mmol·L-1(0.5-2.2mmol·L.-1);Pyruvate97μmol·L.-1(45-80μmol·L.-1);和乳酸/丙酮酸比例12(6-14)。肌肉活检显示只有非特异性的肌炎变化;没有粗糙的红纤维,夹杂物或线粒体异常和肌纤维完好无损。细胞色素氧化酶和肌磷酸化酶染色是正常的。电子显微镜检查未表明细胞细胞形态异常。尿有机酸谱显示多种二羧酸,包括己二酸(C6)和脱苯甲酸(C8)。怀疑β-氧化缺陷。通过串联质谱法测定血清游离肉碱浓度和酰基肉碱图案。血清酰基碱谱发现非常长链酰基氨基碱(C14:20.26μmol·L-1(<0.08μmol·L-1);C14:10.53μmol·L-1(<0.18μmol·L-1);C16:10.16μmol·L-1(<0.08μmol·L-1)))。整体生化调查结果非常暗示VLCAD缺乏。
通过培养的成纤维细胞中的酶测定证实了VLCAD缺乏的诊断通过[13.c] - 抗性加载试验,其显示vlcad活性的标记降低0.8 nmol·min-1·米格-1蛋白质(正常范围:4.21±0.71 nmol·min-1·米格-1),确认VLCAD水平的缺陷。
27天后,患者从医院排出,常规恢复和正常肌酸激酶水平。3个月后进行的肺功能试验,当患者无症状时,显示典型的吸气和呼气肌弱点模式(强制呼气量82%预测,强制生命能力75%,总肺容量60%,残留量20%,强制剩余容量50%,肺部延伸能力为一氧化碳75%,肺部转移系数为一氧化碳93%,最大近透气69%,最大呼气压力28%和最大吸气压力62%)。
讨论
运动不耐受和复发性肌球囊尿素是常均遗传肌肉疾病的常规呈现特征,具有来自各种代谢缺陷的能量产生的潜在缺陷,包括:碳水化合物代谢(糖酵解/糖溶解障碍)中的缺陷;脂肪酸代谢(β-氧化酶的缺乏,肉碱缺乏综合征和脂肪酸输送缺陷);和电子传输链途径(线粒体缺陷;图1)。这些代谢肌病的临床特征可能因呈现而异。新生儿和婴儿细胞能量产生的缺陷通常会产生严重的多系统疾病,而成人发作疾病通常仅限于肌肉。
对于脂肪酸代谢的障碍,肉毒氨基棕榈酰转移酶(CPT)II缺乏是最常见的缺陷,以及复发性横纹肌溶解的最常见的代谢原因。患者的临床特征是脂肪酸代谢障碍的暗示性,但随后的生物化学测定表明了罕见的VLCAD缺乏的存在。
VLCAD缺乏是线粒体脂肪酸β-氧化的遗传障碍,导致长链脂肪酸的氧化缺陷。在长时间禁食后,身体从碳水化合物切换到脂肪酸的能量产生。在VLCAD缺乏症中,由于无法利用长链脂肪酸,延长禁食和过度劳累使患者倾向于急性代谢失代偿,低钾低血糖,心肌病和横纹肌溶解1。
酶VLCAD是由Izai发现的等等。21992年。这种疾病的遗传是常染色体隐性。酶缺乏的酶缺乏的确切频率尚不清楚,但是,在一个试点群众新生儿筛查计划中,脂肪酸氧化缺陷的发生率,包括短链,中链和非常长的酰基 - 辅酶脱氢酶缺陷发现大约是12,000人中的一个3.。
VLCAD缺乏的表型是异质的,并且可以在新生儿中临床分为三种形式:1)肝功能障碍和心肌病;2)后期在婴儿期或儿童时期与低钾低血糖发作和肝功能障碍;3)在具有复发性横纹肌溶解的青少年或成人中。大多数患者具有严重的儿童形式,其特征在于心肌病,早期发作和结果差。在受影响的患者中,由于无法代谢脂肪酸并产生酮体,当葡萄糖和糖原储存耗尽时,低钾低血糖发生。本研究中的患者属于稀有症状后的罕见成人发作型,并且在劳累后和没有低血糖症4.那5.。受影响的个体通常存在于肌肉无力或剧烈运动后通过深色尿液。对于本患者,已排除横纹肌溶解的常见原因,包括肌肉损伤,败血症,药物,毒素和炎症性肌病。肌肉活组织检查是非特异性的,显示出非特异性次要的近视特征,具有正常的细胞色素氧化酶和肌磷酸化酶染色,并且电子显微镜下没有异常细胞细胞形态。禁食乳酸是正常的。因此,鉴于经常性肌肉疼痛(横纹肌)和暗尿(Myoglobinuria)延长施加或禁食后,怀疑脂肪酸代谢障碍。
急性病患者中代谢肌病的初始诊断方法是尿二羧酸的测定。患者中的图案与脂肪酸β-氧化缺陷相容。血液酰基碱图案随后有助于区分β-氧化缺陷的亚型。在目前的情况下,血清和尿液生物化学发现很大程度上是VLCAD缺乏的暗示。培养的成纤维细胞和遗传突变分析中的酶测定证实了诊断。VLCAD的缺陷基因已位于带P11.2和P11.13105之间的染色体17的短臂6.。VLCAD缺乏的分子基础与文献中报告的> 80个突变非常多样化7.那8.。
脉搏症患者的呼吸衰竭程度肌肉弱点罕见。已经在少数CPT II缺乏患者中描述了,但患有VLCAD缺乏的患者之前尚未描述呼吸衰竭。这似乎是这种疾病的第一个报告,呈现为急性高态呼吸急性呼吸衰竭。患者迟到的医院呈现可能会占其发生。当患者从急性发作完全回收患者时获得的肺功能数据证明了呼吸肌弱症的典型发现。这提供了脂肪酸代谢缺陷患者慢性亚临床呼吸肌无效存在的第一种证据。这种情况也是中国患者在文学中报告的第一个VLCAD缺乏。目前作者认为,凭借适当的生化,酶促和突变分析,旨在通过适当的生化,酶促和突变分析支持,认为将来会有更多的案例。
总之,当青少年或成人患者具有无法解释的急性呼吸衰竭,肌肉疼痛和横纹肌分解时,应考虑脂肪酸β-氧化的遗传缺陷,这是通过长时间运动和禁食引起的。
- 已收到2005年11月25日。
- 公认2006年2月17日。
- ©ers Journals Ltd











{kind=link}
{kind=link}