





为了有效地靶向炎症过程,有必要了解真核细胞内炎症反应是如何产生和维持的。因此,本文将首先集中于驱动炎症基因表达的信号转导途径和分子机制,然后反驳一些已发表的假说,试图解释糖皮质激素的抑制作用。目前的工作模型包括,它解释了糖皮质激素的作用,并额外指出了如何阻止炎症基因表达的替代或合作方式。
简介
尽管炎症过程会产生不同的疾病,这取决于所涉及的炎症组织或器官,但所有这些疾病都有共同的方面或共同的细胞过程,如应激信号通路的激活和伴随的炎症细胞因子的产生。为了从“分子”水平了解炎症过程,为开发新型抗炎药物设计更具体的分子靶点,作者课题组对驱动炎症基因表达的信号转导途径和基因调控机制进行了研究。特别是,他们研究了小鼠成纤维细胞中白细胞介素(IL)‐6和其他相关基因启动子的诱导,以响应炎性刺激,如肿瘤坏死因子(TNF),并发现转录因子核因子(NF)‐κB对这些基因的转录诱导至关重要。该因子是一种异二聚体复合体,由两个亚基组成,一个p50脱氧核糖核酸(DNA)结合亚基和一个p65 DNA结合和转录活性亚基。在细胞静息状态下,NF‐κB复合体通过抑制分子IκB在细胞中储存并保持非活性。由于各种炎症刺激,这种抑制剂成为完全靶向降解的对象,释放NF - κB复合体,允许其迁移到细胞核,并与存在于各种细胞启动子中的NF - κB响应序列结合。这可能导致必要的辅助因子的招募,并将引起NF‐κ b驱动基因的诱导,与基础转录机制一起。此外,IκB‐α基因(IκB抑制剂的一种亚型,在炎症刺激的情况下具有明确的靶向作用)在其启动子序列中也包含NF‐κB响应元件,这导致NF‐κB的上调,并重新合成抑制分子,现在可以捕获和结合新合成的和先前释放的NF‐κB。这是一种自动调节回路(负反馈)机制,保持NF‐κB水平的平衡,并严格控制基因转录。
炎症基因的双重激活途径
炎症基因启动子是复杂的启动子序列,由许多不同的响应序列组成;然而,研究发现,在众多转录因子结合的可能性中,转录因子NF‐κB(唯一的)负责对TNF应答的基因诱导。其他启动子结合因子(如激活蛋白‐1,环磷酸腺苷响应性元素结合蛋白,CCAAT增强结合蛋白,等。)与NF - κB协同作用,共同形成招募额外核非DNA结合辅因子(辅激活因子)的必要平台,辅激活因子决定炎症基因表达的一般“水平”,而不是炎症基因的诱导。总之,诱导因子NF‐κB可以被视为炎症基因诱导的最终开关,但在启动子水平上,它与邻近的启动子结合的转录因子和相关的非dna结合的核共激活因子协同作用,以驱动炎症基因的表达。启动子定义的和DNA结合的转录因子的整个相关结构,连同专门招募的非DNA结合的核辅因子被称为“增强体”,这种结构被发现是基因转录的必要步骤1.
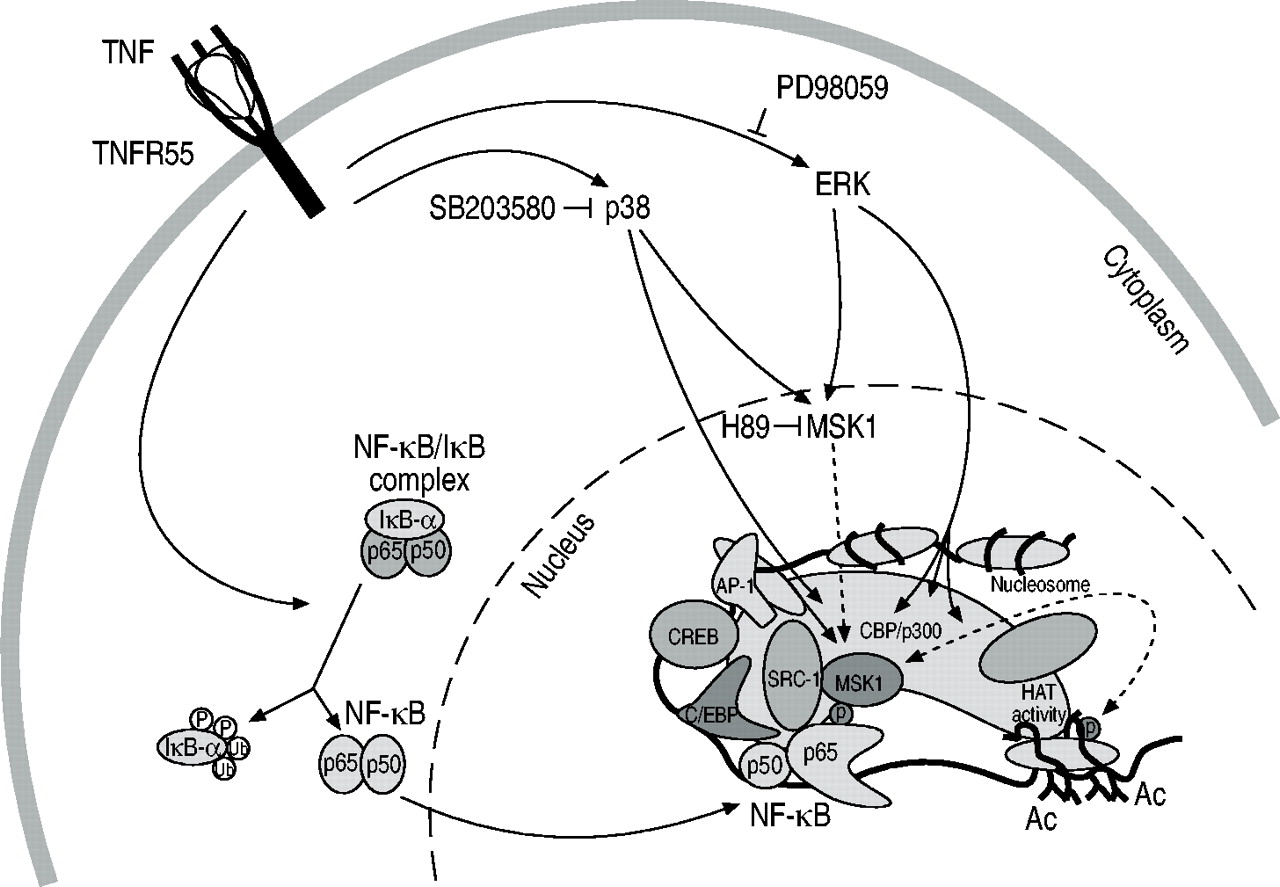
用细胞外信号相关激酶(ERK)和/或p38丝裂原激活蛋白激酶(MAPK)通路的抑制剂预处理TNF‐刺激的细胞,可消除炎症基因表达,但对细胞质NF‐κB激活无影响,也不影响NF‐κB/DNA结合。因此,我们假设,结合复合体(即p65亚基)的转激活能力可能被这些类型的抑制剂干扰,而不是NF‐κB的激活。即。通过阻断磷酸化级联2.ERK激酶和p38 MAPK都不能直接磷酸化NF‐κB p65亚基;有丝分裂原和应激激活蛋白激酶(MSK)1被发现是p65磷酸化的执行酶。事实上,这个激酶在这两个MAPK通路中都位于ERK和p38的下游,是一个纯核激酶,整合了细胞生长或应激的上游传入信号。因此,阻断这种特定的激酶就等于抑制炎症基因的表达。
作者的团队也有明确的证据表明MSK1与NF‐κB p65相关,此外,MSK1/p65的关联发生在IL‐6启动子水平上。然而,这只发生在细胞受到TNF刺激时,而不是在静息状态下,也不是在MAPK通路或MSK1被阻塞时;这就是说MSK1/p65的相互作用和随后的磷酸化是一种特异性的、炎症刺激依赖性事件。MSK1使特定的残基磷酸化(即。NF‐κB p65亚基内的Ser 276)。这个氨基酸位置是一个非常关键的残基,因为突变到另一个氨基酸完全废除了NF‐κB p65的转录效力。事实上,p65分子在这个特定的位点发生突变后,就不再能够促进转录能力增强体的形成,因此也就不能驱动基因表达。
MSK1不是一个新的激酶,但被描述为特异性磷酸化核小体结构内组蛋白的突出肽尾。这些尾部的修饰目前被认为是染色质内定义其进一步任务的“局部编码”过程。在这个假设中,导致弛豫的染色质修饰与增强的基因表达并行。因此,MSK1似乎有双重功能,即。作为一种特异性的NF‐κB p65激酶,以及染色质弛豫的重要信号中间体3..
总之,目前的炎症基因诱导模型涉及双信号通路(图1)⇓).一方面,转录因子NF‐κB从其抑制分子中释放出来,能够与各种炎症基因启动子结合。另一方面,MAPK通路同时被激活,并导致MSK1在关键氨基酸残基上对NF‐κB p65亚基进行特异性磷酸化,这对增强小体的构建至关重要,而在炎症启动子区域被募集的MSK1也会磷酸化周围的染色质尾,有助于局部染色质弛张,促进基因转录。
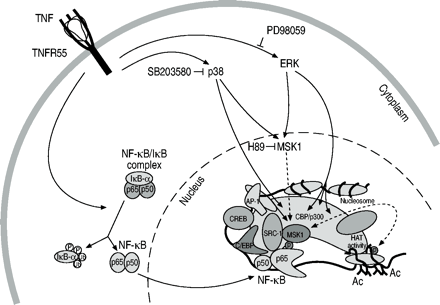
肿瘤坏死因子(TNF)激活炎症基因表达通过双重信号通路。图中显示,在触发膜结合的TNF受体R55后,转录因子复合物核因子(NF)‐κB的细胞质激活转移到细胞核,并在白介素6启动子中的NF‐κB响应元件上占据其位置,旁边是其他已经构成结合的转录因子。同时,有丝分裂原激活的蛋白激酶p38和细胞外信号相关激酶(ERK)也被TNF诱导激活,然后激活它们共同的核底物,有丝分裂原和应激激活蛋白激酶(MSK)1,它在关键的残基磷酸化NF‐κB p65,使其成为环腺苷单磷酸反应元件结合蛋白(CREB)结合蛋白(CBP)/p300和其他核共激活因子的相互作用伙伴。组蛋白尾巴被MSK1磷酸化,并被核共激活物乙酰化,以放松染色质和增强基因表达。AP:激活蛋白;SRC:类固醇受体共激活剂;C/EBP: CCAAT增强结合蛋白;p/CAF: p300/CBP激活因子;组蛋白乙酰转移;Ac:乙酰基。
糖皮质激素对基因的抑制
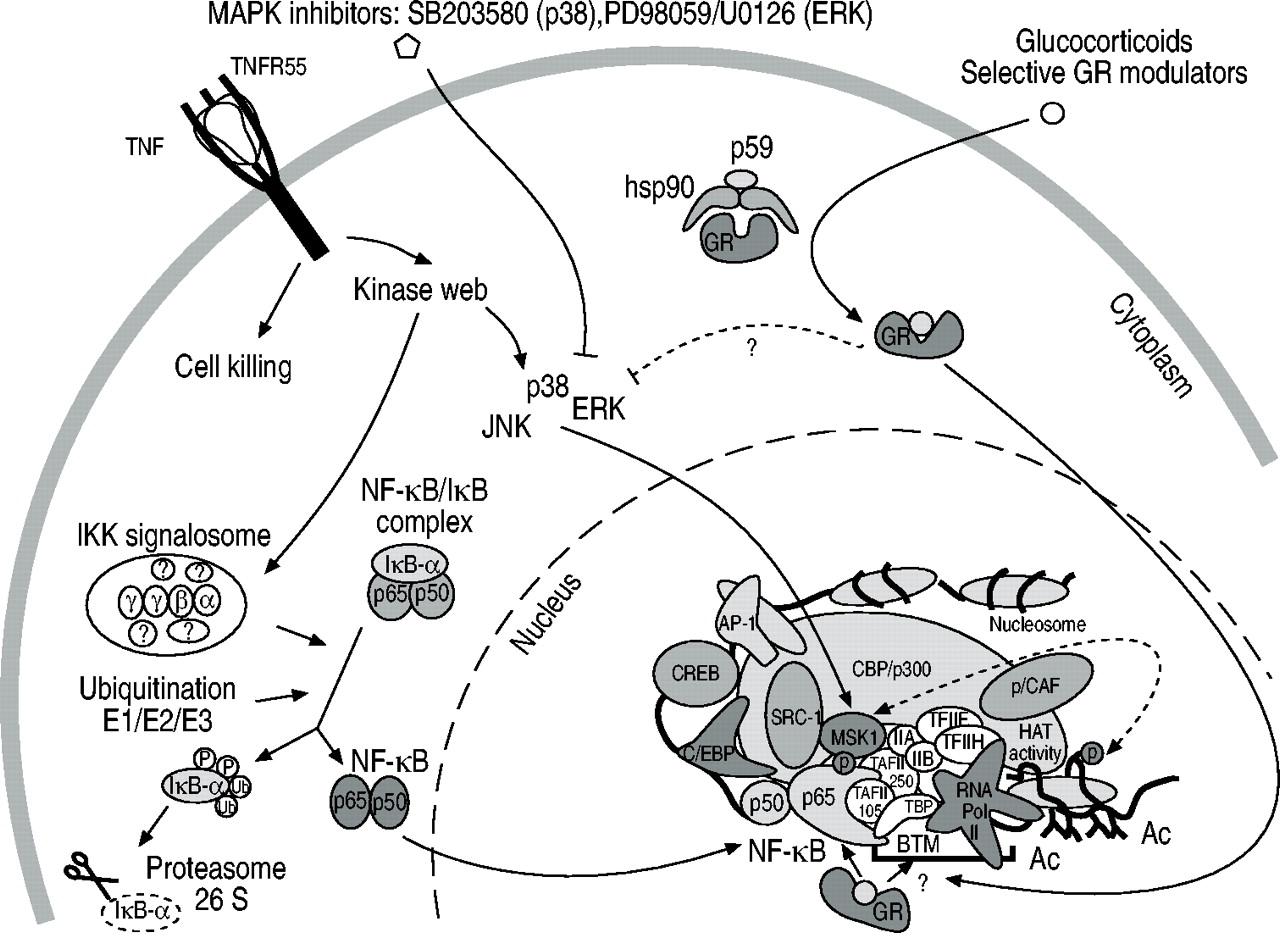
糖皮质激素被认为是对抗炎症的有效药物,并被广泛用于这一目的。这些亲脂性化合物可以穿透细胞膜并与特定的受体分子结合,即糖皮质激素受体(GR)。受体从一个巨大的细胞质蛋白复合物中释放出来,并作为配体载体迁移到细胞核中,在那里它成为激活的转录因子。因此,它将与各种基因启动子中所谓的(主要是回文)糖皮质激素响应序列元件结合,并与其他因子协作,建立基因表达所必需的增强体结构。然而,糖皮质激素的抗炎潜力与激活GR的基因诱导活性无关,而被认为是由炎症、即。NF‐κ b驱动基因表达。一些作者指出了糖皮质激素的潜力,即。上调NF κB -抑制分子IκB - α基因的表达4,5但是,本作者的团队没有在成纤维细胞或内皮细胞中发现GR的这种调节作用,也没有观察到糖皮质激素治疗后NF‐κB结合的减弱。相反,他们证明糖皮质激素对NF - κB活性的抑制发生在没有基因转录和蛋白质合成的情况下,并且完全发生在细胞核中6.其他研究人员认为核GR是激活的NF - κB在基因启动子上组装必要增强体的竞争对手,导致NF - κB基因转录明显下降7,8.作者小组发现事实并非如此,并确定即使在GR抑制的条件下NF‐κB仍与增强体伙伴保持联系。此外,该假设提出了基因抑制的一般方式,并没有解释糖皮质激素对炎症的特异性9,10.糖皮质激素对染色质修饰没有影响,也没有发现对上述磷酸化级联的干扰。这就是说,必要的“MAPK→MSK1→p65 transactivation”通路不受糖皮质激素的影响,因此可能包含抗炎策略的替代靶点,无论是否与糖皮质激素联合治疗。
作者目前的工作模型提出GR可特异性干扰NF‐κB p65与基础转录机制之间的相互作用平台,其中基因抑制的特异性由相关因素的设置和空间组织保证,如由炎症基因启动子的主要序列决定。
结论
总之,核因子- κB驱动的炎症基因的转录激活的双重信号通路的发生和必要性已经被证实,其中炎症基因启动子的特异性可能以某种方式受到伴随的染色质弛豫的调节或指示。糖皮质激素抑制炎症基因后,转录活性复合体和染色质结构不受影响;然而,激活的糖皮质激素受体似乎干扰核因子‐κB p65与基础转录复合体之间的接触,从而以启动子结构特异性的方式下调基因表达11(图2⇓).
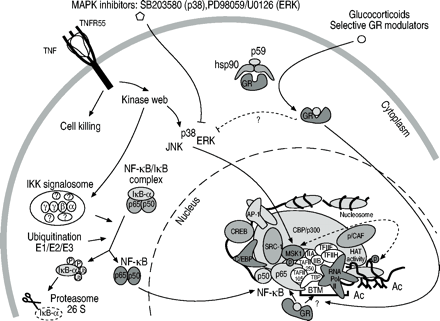
糖皮质激素与糖皮质激素受体(GR)结合,糖皮质激素受体进入细胞核,以启动子结构的特定方式抑制炎症基因表达。该图显示了与图1所示相同的肿瘤坏死因子(TNF)驱动的基因激活途径⇑.然而,添加糖皮质激素后,GR从细胞质蛋白复合物中释放并转移到细胞核中,在那里,在基因抑制的情况下,GR特异性地与核因子(NF)‐κB p65和基础转录机制(BTM)之间的激活界面相互作用,从而以启动子特异性的方式抑制基因表达。注意GR可能不会干扰丝裂原激活蛋白激酶(MAPK)级联。AP:激活蛋白;CREB:环磷酸腺苷反应性元素结合蛋白;C/EBP: CCAAT增强结合蛋白;ERK:细胞外信号相关激酶;JNK: Jun N‐末端激酶;RNA:核糖核酸;SRC:类固醇受体共激活剂; p/CAF: p300/CREBbinding protein (CBP) activating factor; HAT: histon acetyl transferation; Ac: acetyl; TBP: TATA‐binding protein.
- ©ERS期刊有限公司










{kind=link}
{kind=link}
{kind=link}
{kind=link}