





文摘
背景特发性肺动脉高压(IPAH)是一种罕见的疾病遗传可能性高。尽管一些诱发基因与IPAH,遗传病因学大量IPAH例仍然未知。
方法我们进行了一次exome-wide基于基因的负担分析两个独立的病例对照研究,包括331 IPAH病例和508控制。功能进行了评估分析在蛋白质生物合成和基因突变的影响函数。
结果人类骨形态形成蛋白基因编码9 (BMP9)被确定为一个新的基因位点与IPAH显示exome-wide协会发现队列(或18.8;p = 1.9×10−11)。本协会认证的独立复制组(p = 1.0×10−5)。总的来说,罕见的编码突变BMP9该基因发生在6.7%的情况下,排名第二BMPR2,包括合计2.7×10的重要性−19(或21.2)的风险。有趣的是,患者BMP9突变比那些没有等离子BMP9水平较低。功能研究表明BMP9突变导致BMP9分泌减少和肺动脉内皮细胞的抗凋亡能力受损。
结论我们确定了BMP9作为一个IPAH罪魁祸首的基因。
文摘
BMP9是一个新的罪魁祸首基因IPAH排名第二BMPR2。罕见的有害突变BMP9导致BMP9分泌减少,损害BMP9函数,占IPAH病例的6.7%。http://ow.ly/h0tS30mXr8j
介绍
特发性肺动脉高压(IPAH),占35 - 67%的肺动脉高血压(PAH)情况下,是一种罕见但致命的疾病遗传高(1- - - - - -4]。的遗传基础IPAH已经被广泛的研究。罕见的编码突变骨形态形成蛋白受体2型(BMPR2)在∼70%的家族血缘的多环芳烃和15 - 20%的零星IPAH情况下(5- - - - - -7]。较少,存在重大影响的罕见突变基因编码caveolin-1 (CAV1),钾通道二端口域亚K成员3 (KCNK3),SMAD家庭成员1 (SMAD1),SMAD家人9 (SMAD9),T-box 4 (TBX4)和骨形态形成蛋白受体1型b (BMPR1B)[8- - - - - -13]。此外,激活素受体1型(ACVRL1)和endoglin (英格)被确定为基因位点与发展的多环芳烃和遗传性出血性毛细血管扩张(HHT) [14,15]。biallelic隐性突变的真核翻译起始因子2α激酶4 (EIF2AK4)最近被证明是肺部静脉阻塞疾病的主要原因和/或肺毛细管haemangiomatosis、临床和病理不同形式的多环芳烃(16]。10基因的突变都只能解释一小部分(IPAH∼20%)情况下,建议额外IPAH基因必须存在(17]。
规范化方法来识别IPAH易感基因涉及家庭遗传的使用。到目前为止,数据生成的多基因多环芳烃家庭常用的揭示突变在这些已知的PAH基因之一。IPAH的稀缺性使得更难识别新IPAH-related基因通过针对大量的例nonbiased全基因组筛选。此外,PAH源于一种相互作用在许多不利的因素,包括,但不限于,毒品、环境因素、年龄、性别和种族背景。因此,基因型数据不会唯一临床异质性的混杂因素。与上述局限性,大规模基因标本的分析和定义良好的IPAH情况下结合最先进的方法是急需确定其他IPAH易感性位点。
新一代测序的数据分析(上天)使小说致病基因的识别。这里,我们旨在识别IPAH诱发基因通过执行两阶段总会在零星IPAH情况下在中国参加两个中心> 10年。而先前确定的程度PAH基因(例如BMPR2)验证,基于基因的负担分析筛选IPAH易感基因。
方法
可以找到所有方法的详细描述补充材料。
研究设计
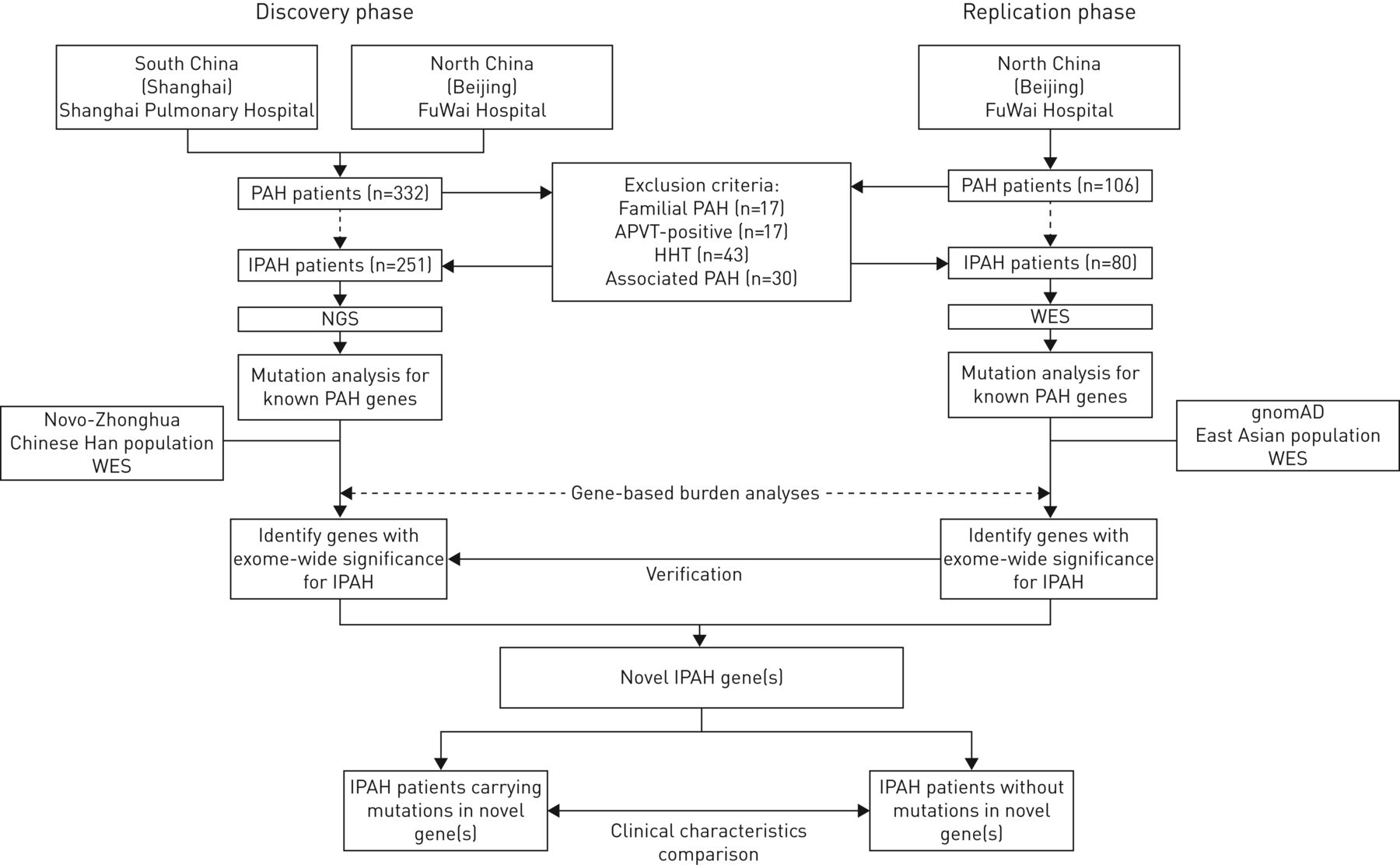
IPAH从两个国家肺动脉高压患者转诊中心在中国:上海肺科医院上海同济大学(患者主要来自中国南部)和中国医学科学院阜外医院在北京(患者主要来自中国北部)。这项研究包括两个阶段:“发现”和“复制”阶段。所有IPAH病例组由门店和筛查罕见变异在10之前报道PAH基因(BMPR2,ACVRL1,英格,SMAD1,SMAD9,TBX4,BMPR1B,CAV1,KCNK3和EIF2AK4)。基于基因的负担分析当时执行的比较情况下对控制的外显子组数据来识别小说IPAH易感基因。这些假定的基因识别发现队列随后被评估在复制队列。基因型与临床表型之间的关系进一步分析使用数据集相结合。研究协议制度审查委员会批准在上海肺科医院、阜外医院。显示了研究设计图1。
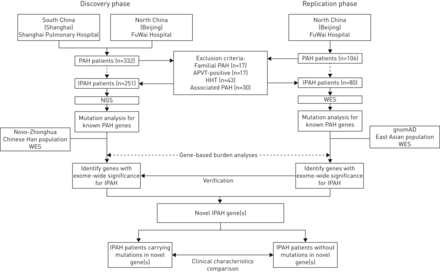
研究设计的流程图。多环芳烃:肺动脉高血压;IPAH:特发性肺动脉高血压;APVT:急性肺vasoreactivity测试;遗传性出血性毛细血管扩张症:遗传性出血性毛细血管扩张;门店:下一代测序;韦斯:whole-exome测序;gnomAD:基因聚合数据库。
统计分析
结果报告为百分比,中位数(四分位距(差))或均值与标准差,表示。数据分布的正常使用Kolmogorov-Smirnov测试评估。卡方测试或确切概率法应用于比较定性变量和基因型和等位基因频率。对于定量变量,统计学意义决定使用一个未配对t检验或单向方差分析。的Mann-Whitney紫外线测试或克鲁斯卡尔-沃利斯检验进行分析非正态的分布变量。统计测试都是双尾。结果被认为是p < 0.05的显著水平。所有与PASW统计分析版本18.0(美国SPSS,芝加哥,IL)。
结果
研究人群
共有331名患者IPAH是从两个国家转诊中心(招募图1)。发现组,122例儿科患者(15)是从上海肺科医院和招募129例(33儿科病人)是从阜外医院招募的。复制队列,80事件例(9儿科患者)在阜外医院登记。人口统计学和临床特点的两个IPAH军团中描述表1。
的识别BMP9作为一个IPAH易感性基因
在发现阶段,我们36 IPAH基因分型情况下使用全基因组测序(WGS),另215例使用whole-exome测序(韦斯)。WGS和韦斯覆盖率达到平均37 - 128倍的目标,分别。超过92%的有针对性的基地被测序读深度超过20倍(补充表S1)。
我们首先分析了罕见变异和突变的数据在10之前报道PAH-predisposing基因。罕见的杂合突变BMPR2被发现在49例(19.5%)。对于其他PAH基因风险,我们确定了有害的罕见变异ACVRL1(15例(6%)),TBX4(10例(4%)),SMAD1(2例(0.8%),BMPR1B(1例(0.4%),KCNK3(1例(0.4%)SMAD9(1例(0.4%))。庇护biallelic 4例(1.6%)EIF2AK4突变:两种情况下进行纯合突变和两种情况进行的杂合突变(一个错义和一个转移在一个案例中,和其他两个错义突变)。8例(3.2%)有双重的杂合突变或罕见变异在两个不同的PAH基因风险。总的来说,罕见变异和突变被发现在91名患者,对应于所有病例的36.3% (补充表S2)。所有的罕见变异和突变的细节10 PAH基因提供了补充数据集S1。
为此,所确定的罕见变异和突变基因已知IPAH证实发现群体的遗传背景有关。然后,我们进行了基于基因的负担分析利用外显子组251例和1884年的数据控制(补充图S1)。同质人口结构的进一步分析表明,所有病例和控制集群对象的中国血统在1000基因组数据集(www.1000genomes.org)(补充图S2)。总共13 318个基因有超过251年的一个案例IPAH患者潜在的有害的基因变异的主导模式测试。正常化后(p < 3.8×10多个测试−6占13 318个基因检查)后,只有三个基因(BMPR2,BMP9(编码骨形态形成蛋白9)ACVRL1)有一个exome-wide IPAH显著富集的突变情况下与控件(相比表2和补充图S3)。值得注意的是,除了证据确凿的PAH使役动词BMPR2和ACVRL1,BMP9将是一个新的基因显示与IPAH exome-wide协会。突变BMP9被发现在6.8%(17 251)的情况下,0.4%(1884 7)的控制(或18.8,95%可信区间7.4 - -53.9;生p = 1.9×10−11,Bonferroni纠正p = 2.6×10所示−7)。在主要的编码模式下,BMP9排名第二的旁边BMPR2(表2和补充图S3)。具体地说,BMP9突变被发现在5.7%(7 122)和7.8%(10 129)从上海肺科医院、阜外医院的病例,分别为(表1)。的分布BMP9突变的两个中心没有显著差异(p = 0.53,卡方测试)。
为了进一步验证的风险影响BMP9,我们执行80年独立复制队列韦斯IPAH病例。罕见变异和基因突变在PAH-related 37例(46.3%)病例中标识(总结补充表S2;列出所有的罕见变异和突变补充数据集S1)。使用相同的管道遗传负担分析,我们比较了80例参考控制外显子组测序数据的外显8624年东亚人的基因组聚合数据库(gnomAD-EAS;http://gnomad.broadinstitute.org)。BMPR2,BMP9和ACVRL1再次确认为的三大疾病有关的基因复制队列(表2)。我们确定了五个额外的(6.3%)BMP9的杂合突变(表1和补充表S3)。的患病率BMP9突变的参考控制明显高于gnomAD-EAS(原始p = 1.0×10−5)。在合并后的发现和复制数据集,突变BMP9被发现在6.7%(22 331)和0.3%(34 10 508)的病例和控制,分别由2.7×10的组合意义吗−19(或21.2,95%可信区间11.7 - -37.6)。的患病率BMP9突变被发现和复制组(6.8%之间的可比性与6.3%)。因此,这强烈表明,两阶段分析BMP9轨迹IPAH的发病机制是一种新型的贡献者。
BMP9突变特征
在331 IPAH患者中,我们确定了22例21个不同的罕见的杂合突变BMP9(补充表S3)。这些突变传遍BMP9(图2一个和b)。所有的BMP9突变经Sanger测序(补充图S4)。没有一个BMP9突变携带者有任何有害基因突变在10之前报道的多环芳烃。20这些BMP9突变是缺席超过6512外显或基因组,包括301名中国样本1000人基因组计划,1884年Novo-Zhonghua控制和4327东亚人外显子组聚合财团数据库(ExAC-EAS;http://exac.broadinstitute.org)。只有一个突变(p.G322R)被确定在这些数据库中,患病率为0.0001 -0.0002 (补充表S4)。的22个BMP9突变携带者,功能丧失(LOF)突变被发现在6例(27.3%),包括三个无意义突变,两个移码和一个突变,会影响规范转化开始网站(补充表S4)。在这两个参考数量,没有这样的BMP9突变被发现。的患病率BMP9LOF突变IPAH例(6例(1.8%)明显高于两个参考人群(p = 1.1×10−5Novo-Zhonghua和p = 2.5×10−9gnomAD-EAS控制)。关于15BMP9错义突变,在网上分析表明,这些突变是有害的(补充表S4)和位于不同物种中高度保守的区域(图2 c)。
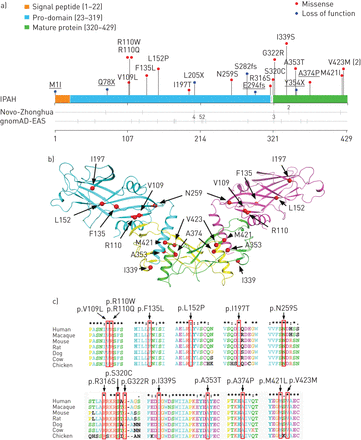
骨形态形成蛋白9 (BMP9)突变在特发性肺动脉高压(IPAH)。gnomAD-EAS:基因组数据库东亚聚合。突变的空间分布BMP9发现患者IPAH以及变异的分布在健康控制。突变复制队列中标识下划线。的受试者携带数量相同的突变。11 b)分布BMP9错义突变被发现在IPAH病人显示在BMP9的晶体结构,在生长因子叠加二聚体。pro-domain丝带颜色是蓝色和紫色。颜色是黄色和绿色的成熟的增长因素。c) ClustalX (www.clustal.org)的氨基酸序列比较显示了15个错义的高保护BMP9在不同的物种突变。突变红框所示。
的临床特点BMP9突变携带者
补充表S3显示了临床、功能和血液动力学的特征的22个IPAH病人携带的各种BMP9突变。雄性和雌性都受到影响,显著的性别偏见是明显的女性:男性的比例4.5:1。7例(31.8%)在随访期间死亡。
我们扩大了的分析BMP9基因型和表型结果合并后的数据集。与案件有或没有BMP9突变,年龄和血液动力学(没有明显差异补充表S5)。作为BMP组的成员将增长factor-β总科蛋白质,BMP9作为429 -氨基酸合成前体蛋白(pre-pro-BMP9)和加工成pro-BMP9 (100 kDa)和成熟BMP9 (kDa 25日)(补充图S5)。活动形式的BMP9循环血液中,调节血管的基调是至关重要的18]。调查是否BMP9突变影响BMP9的表达水平,分析了等离子体BMP9水平IPAH病人携带BMP9突变(n = 19)、年龄和sex-matched健康对照组(n = 87)、和IPAH病人BMP9突变(n = 38) (补充表S6)。三组之间的BMP9水平明显不同(p < 0.0001,克鲁斯卡尔-沃利斯检验)(图3)。与健康对照组相比,等离子体IPAH患者缺乏BMP9水平低BMP9突变(中值(差)19.8 (15.2 - -43.4)与36.2 (20.8 - -60.7)pg·毫升−1;Mann-Whitney测试,p = 0.005)。重要的是,BMP9水平更低BMP9突变携带者与患者相比没有突变(中位数(差)12.1 (8.6 - -21.8)与19.8 (15.2 - -43.4)pg·毫升−1;p = 0.002, Mann-Whitney测试)(图3)。这些结果表明,BMP9突变可能抑制BMP9合成和/或分泌,导致循环BMP9水平下降可能IPAH的温床。
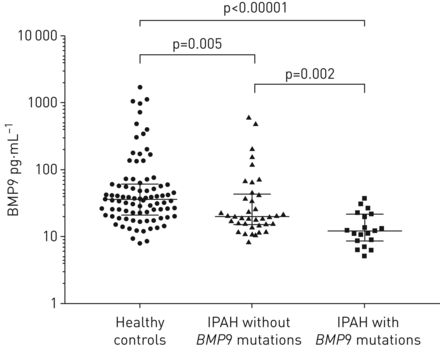
骨形态形成蛋白9 (BMP9)水平在健康对照组(n = 87)、特发性肺动脉高压(IPAH)患者BMP9突变(n = 38)和IPAH患者BMP9突变(n = 19)。BMP9 IPAH病人没有减少BMP9突变和最低BMP9突变携带者。并给出了数据作为单独的数据点用中位数和四分位范围。统计分析两组之间使用Mann-Whitney测试执行。
功能的研究BMP9突变
调查造成有害的结果BMP9突变,我们选择了六个突变功能评估基于临床表现(补充图S6)。三个突变(V109L S282fs和V423M)被选中,是因为这些突变患者疾病的早期发病。其他三个突变(R316S S320C和A353T)进行了研究,因为突变携带者有严重的临床表现,诊断后在短时间内下降(补充表S3)。免疫印迹显示,野生型(WT)和五个错义质粒(V109L、V423M R316S, S320C和A353T)表达了类似的水平pro-BMP9蛋白质在细胞溶菌作用(补充图S7)或在上层清液从人类胚胎肾HEK293E细胞(图4一右面板)。然而,明显降低在成熟BMP9观察BMP9突变体(图4一左面板)。截短突变S282fs,无论是pro-BMP9还是成熟BMP9可以检测到(图4一和补充图S7)。这些结果证实了ELISA试验(图4 b)。成熟的BMP9水平显著降低HEK293E文化上层清液的细胞转染V109L, R316S S320C,和S282fs几乎检测不到,A353T V423M。然后我们使用条件媒体V109L - R316S——S320C-transfected细胞检查他们的能力在BMP信号通路激活(图4 c)。BRE (BMP响应元素)荧光素酶活性测定表明,R316S展出活动与WT BMP9相比下降。此外,预处理WT BMP9条件媒体保护肺动脉内皮细胞(PAECs)细胞凋亡诱导的肿瘤坏死factor-α环己酰亚胺的存在。然而,所有的三个突变体仍未能阻止PAEC细胞凋亡(图4 d)。
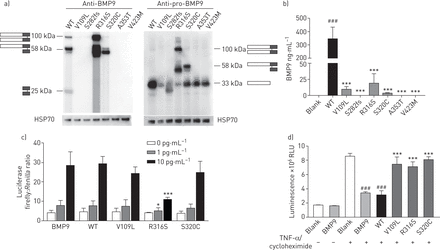
骨形成蛋白的功能研究9 (BMP9)突变。WT:野生型;HSP:热休克蛋白;RLU:相对光单位;肿瘤坏死因子:肿瘤坏死因子;PAEC:肺动脉内皮细胞。一)人类胚胎肾HEK293E与突变细胞转染BMP9显示出不同的表达模式与WT。HEK293E与WT或细胞转染BMP9突变质粒。免疫印迹的上层清液是收获一种抗体特定成熟BMP9或pro-BMP9。WT BMP9蛋白质分离三个乐队对应pro-BMP9 (100 kDa),部分加工pro-BMP9 (58 kDa)和成熟BMP9二聚体(kDa 25日)。b) ELISA试验表明,成熟的分泌BMP9显著低于HEK293E细胞转染的上层的突变BMP9比WTBMP9。方差分析。* * *:p < 0.001 WT相比;# # #:与空白相比p < 0.001。c)的错义BMP9突变R316S引起显著降低正好与WT (BMP响应元素)活动BMP9。与人类co-transfection后激活素受体激酶1,BRE-luciferase和强RenillaC2C12细胞治疗与WT或突变BMP9上层清液1和10个pg·毫升−1,分别。商业BMP9重组蛋白作为积极的控制。方差分析。*:p < 0.05 WT相比;* * *:与WT.相比p < 0.001 d)BMP9减毒突变PAECs WT BMP9的抗凋亡作用。PAECs与WT预处理或突变BMP9上层清液ng在5毫升−1。的突变体的影响BMP9PAEC凋亡的评估是通过测量caspase-3/7活动后刺激TNF-α和环己酰亚胺。方差分析。* * *:p < 0.001 WT相比;# # #:与空白相比p < 0.001。
讨论
在目前的研究中,我们使用门店识别BMP9是一种新型IPAH易感性基因。罕见的BMP9突变赋予> 21-fold IPAH的几率较高,排名第二BMPR2。功能的研究证实了这些BMP9突变影响循环BMP9水平IPAH病人。此外,这些突变体造成有害的影响在培养细胞蛋白质表达和功能。综上所述,这些研究结果强烈表明,突变BMP9病因学的多环芳烃。
门店涉及外显子组测序导致小说遗传基因的识别多环芳烃(HPAH)在家庭拥有多个影响个人10,11),然而,挑战依然存在,当将该技术应用于确定新的基因IPAH无关的主题之一。困难来自大型均匀病人群的招聘。在最近的研究中,我们从两个专家中心招募病人IPAH一致的诊断程序。几乎所有家庭多环芳烃(FPAH)患者有已知的PAH基因,基因突变和急性肺vasoreactivity阳性反应者不同于IPAH患者的发病机理和遗传病因学(19]。因此,我们将这两个子组的患者排除在IPAH群体更好地解读IPAH的遗传基础。我们选取57儿科IPAH病人(意味着±sd9.9±5.5岁),274名成人患者(平均±sd31.8±7.7岁)和排除病例> 60岁,导致总平均年龄28岁。此外,所有受试者参加本研究正式注册为中国汉民族。因此,严格的选择标准,精确的临床表型与相对年轻和齐次军团在当前的研究中会产生令人信服的数据IPAH-predisposing基因。因此,我们的数据也证明了先前确定的10 PAH基因的突变率较高(38.7%)与19.9%),BMP9(6.8%与1.1%)在当前的研究中比在欧洲群G英国皇家空军等。(20.]。
不同人群和种族之间的遗传背景的差异导致独特的特定基因参与罕见疾病的倾向。例如,因子V莱顿,静脉血栓栓塞的重要危险因素,平均等位基因频率的欧洲人口的4.4%,但几乎没有东亚人口(21]。因此,确认公认的独立军团之间的罕见疾病基因在不同的大洲是至关重要的识别易感基因在疾病的病因学中的作用。最近,克英国皇家空军等。(20.)执行欧洲白种人的WGS IPAH / HPAH病人和报道的一个重要群体基因突变在四(ATP13A3(腺苷三磷酸酶13 a3),AQP1(aquaporin-1),SOX17(SRY-box 17)BMP9)。在这些基因中,只有BMP9在我们的研究中进行了验证。没有其他的三个基因(ATP13A3,AQP1和SOX17)结合群体达到统计学意义(补充表S7)。并给出了三个基因的突变补充数据集S2。此外,在21BMP9突变描述在目前的研究中,只有两个BMP9突变之前被报道(20.];剩下的19个是新发现的突变。综上所述,我们的结果不仅补充之前报道的欧洲数据由G英国皇家空军等。(20.),但也加强的关键作用BMP9突变的遗传病因学IPAH。
BMP9(也称为生长和分化因子2)主要是表现在肝脏和既定分泌进入血液循环。通过绑定ACVRL1(骨形态发生蛋白1型受体)和BMPR2(骨形态发生蛋白2型受体),BMP9起着重要的作用在调节血管生物学和血管生成18]。因为ACVRL1和BMPR2肺血管内皮细胞中高度表达,BMP9循环的最优水平是至关重要的维持静止的肺血管22]。在这里,我们提供令人信服的证据表明BMP9是一种多环芳烃通过exome-wide易感性基因研究。所有的21BMP9突变鉴定是非常罕见的,这是符合IPAH的低发病率每年每百万人口(6例)。我们展示了各种各样的BMP9突变可能负面影响BMP9功能,包括合成、分泌、加工和/或激活ACVRL1信号,从而导致haploinsufficiency肺内皮功能BMP的轴。值得注意的是,R316S突变显示一个独特的蛋白表达模式(图4一)和更多的有害影响ACVRL1信号(图4 c)。这些结果可能造成的特殊位置R316S BMP9高度保守的的裂解位点的突变,这可能会干扰蛋白水解乳沟。多环芳烃的发病机制的分子机制引起这些的BMP9突变值得进一步调查。
值得注意的是,ACVRL1排名第三,在所有IPAH-related基因在我们的两个群体,与罕见突变发生在22例。这是以前的研究显示,看似矛盾的ACVRL1是一个主要的致病基因遗传性出血性毛细血管扩张症和多环芳烃与遗传性出血性毛细血管扩张症23,24]。确认我们的结果,医疗记录和审查,以确保所有这些家庭历史ACVRL1突变携带者确实IPAH。ACVRL1突变携带者可能开发多环芳烃和遗传性出血性毛细血管扩张症。78%患者携带ACVRL1从法国PAH突变网络,多环芳烃诊断之前的表现遗传性出血性毛细血管扩张症(25]。此外,由于遗传性出血性毛细血管扩张症几乎完整的外显率在60岁,一些ACVRL1突变携带者可能不年轻时表现出临床证据的遗传性出血性毛细血管扩张症。考虑到年轻的年龄ACVRL1突变携带者确认在我们的研究中(平均25.7(3-50)岁(范围)),这些患者可能不存在遗传性出血性毛细血管扩张症的临床特征。我们会继续关注这些患者确认表型。
最近,一群PAH患者被发现携带不止一个罕见的基因变异,导致双杂合的基因型。例如,克英国皇家空军等。(20.)报道,一个PAH患者进行高度有害的变异BMPR2和SMAD9。在最近的研究中,我们确定了8例携带双突变或罕见变异BMPR2 / ACVRL1,BMPR2 / BMPR1B,BMPR2 / SMAD9,BMPR2 / TBX4和KCNK3 / TBX4。双杂合性的基因突变或变体涉及PAH风险突出PAH固有的复杂性。是否是双变异导致了PAH发病机理和临床结果权证的未来研究。
我们所知,这是第一个报告来识别小说IPAH易感基因在亚洲使用exome-wide研究基于大量的IPAH病人。的识别BMP9突变强烈支持的假设一个障碍BMP信号导致PAH基因的易感性。实验,政府重组BMP9刺激BMP信号在内皮可以预防或逆转多环芳烃在几个基因和nongenetic啮齿动物模型(26]。数据图3表明循环BMP9水平显著降低患者携带BMP9有害的突变。这一发现表明,IPAH病人携带BMP9突变体可以接收BMP9补充疗法作为个性化治疗的一部分。
研究的局限性
警告我们的研究,所涉及的所有患者两组散在病例。由于不可用DNA样本受影响的亲戚、家族性隔离与PAH尚未被证实。是否BMP9变异与FPAH应确认未来的谱系。另一个限制是相对较低的患者数量的复制队列。未来的研究中更大的群组研究中会进一步确认之间的关系至关重要BMP9和IPAH。鉴于循环BMP9 IPAH的水平只测量基线患者或无突变,我们无法测试BMP9的可变性和预后价值随着时间的推移或相关性不同的治疗策略。
结论
使用门店外显子组测序在大同质群体,我们已经证实突变在人类BMP9使IPAH易感性。
补充材料
补充材料
请注意:补充材料并不是由编辑部,编辑和上传已由作者提供。
补充材料:方法、表和数据。erj - 01609 - 2018 - _supplement
补充数据集1:罕见变异和突变在10之前报道PAH基因风险。erj - 01609 - 2018 - _dataset_1
补充数据集2:罕见变异三个候选人PAH基因。erj - 01609 - 2018 - _dataset_2
确认
作者感激地感谢病人和家庭的参与这项研究。
脚注
可以从本文的补充材料www.qdcxjkg.com
利益冲突:X-J。王没有披露。
利益冲突:T-Y。丽安没有披露。
利益冲突:x江泽民没有披露。
利益冲突:自动送料刘没有披露。
利益冲突:S-Q。李没有披露。
利益冲突:r .江泽民没有披露。
利益冲突:W-H。吴没有披露。
利益冲突:j .你们没有披露。
利益冲突:bxcy。程没有披露。
利益冲突:y Du没有披露。
利益冲突:X-Q。许没有披露。
利益冲突:吴y没有披露。
利益冲突:F-H。彭没有披露。
利益冲突:k .太阳没有披露。
利益冲突:y m。毛泽东没有披露。
利益冲突:h . Yu没有披露。
利益冲突:梁没有披露。
利益冲突:J.Y-J。Shyy没有披露。
利益冲突:S-Y。张没有披露。
利益冲突:x张没有披露。
利益冲突:bzcy。京没有披露。
支持声明:拨款支持的工作从北京自然科学基金(7181009,7181009),中国国家重点研发项目(2016 yfc0901502),中国国家自然科学基金(81320108005、81320108005和81320108005),国家杰出青年科学基金(81425002),凸轮医学科学创新基金(2016 - i2m - 1 - 002、2016 - i2m - 4 - 003),和导演基金从心血管疾病国家重点实验室(2017 zr-03 2018 zr-02)。资金信息,本文已沉积的Crossref资助者注册表。
- 收到了2018年8月24日。
- 接受2018年11月29日。
- 版权©2019人队











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}