





到编辑:
先天性角化不良症是一种罕见的外胚层发育不良的遗传性疾病,其典型特征是皮肤色素异常、指甲营养不良和皮肤白斑[1-3.,至少有一种存在于大约80-90%的先天性角化不良病例中。骨髓衰竭是另一个常见的特征,还有各种其他的异常(如。还描述了牙科,胃肠道,神经系统,眼科,肺和骨骼)[1-3.].Dyskeratosis的死亡率的主要原因是骨髓衰竭,肺病和恶性肿瘤是骨髓衰竭[1].已经认识到了三种遗传模式:X链接隐性,常染色体显性和常染色体隐性[1那3.].八种先天性角化不良基因(DKC1中(甜点表达症Congenita 1),TERC.(端粒酶RNA组件),塔特(端粒酶逆转录酶),NOP10.(核仁蛋白10),NHP2.那TINF2.(terf1相互作用核因子2)TCAB1和RTEL1.(端粒延伸解旋酶的调节1))已经被确认,它们的突变占所有先天性角化不良病例的60% [1].在先天性角化不良基因中,突变在TERC.那塔特和DKC1中据报道,已据报道,与家族性肺纤维化和特发性肺纤维化有关,肺纤维化被认为是不良病变的同性恋者的特征之一。然而,尚未澄清其他不良病变中的突变与肺纤维化的关系。据我们所知,这是第一种描述患有肺纤维化患者的疾病患者的案例报告。TINF2.突变。
一位43岁的女性与咳嗽和进步的呼吸困难一起参观了我们的医院。她从未吸过吸烟,患有血栓性贫血,眼泪,Queyrat和不孕症的射伤。她的父亲被诊断出患有增生性贫血,他的整个身体都是色素的。大约2年前,她抱怨咳嗽并咨询了她的个人医生。她的胸部射线照片显示了双侧肺田的漫射网状阴影。她被称为一所综合医院,被诊断出患有特发性肺炎肺炎。因为她的一般情况当时是稳定的,所以她随访1年没有任何特异性治疗。由于呼吸困难逐渐恶化,她被提交给我们的医院,并承认进一步检查。她的身体检查对于她的全身对皮肤色素沉着显着,左眼左眼和细裂纹的眼斑和细裂纹。她的指尖皮肤粗糙,但她的指甲不是营养不良的。 Although no leukoplakia was found in the oral mucosa, she had erythroplasia of Queyrat of the vulva. Laboratory data showed elevated lactate dehydrogenase, transaminases, erythrocyte sedimentation rate and sialylated carbohydrate antigen KL-6 with thrombocytopenia. Chest radiographs demonstrated consolidation and reticular shadows in the bilateral lung fields. Furthermore, chest computed tomography revealed consolidation and reticular shadows in both lung fields, as well as bronchiectasis and cystic shadows in the left lung.
在这一点上,我们强烈怀疑她患有疑难表明同一病。要做出明确的诊断,我们首先检查了TERC.和塔特直接测序基因。然而,在任何一种基因中没有发现突变。Southern印迹分析显示细胞长度短(如图。1A),因此突变TINF2.接下来是探索的。如图所示图1B.,因为直接测序显示了一个n871-874四核苷酸的AGGA缺失TINF2.,她被诊断为具有肺纤维化的患有障碍症的伴有表达病症TINF2.突变。随着她呼吸状况的进展,类固醇脉冲治疗后口服泼尼松龙。然而,她的症状没有得到改善,并出现双侧气胸伴纵隔和皮下肺气肿。开始治疗1年后死于呼吸衰竭。
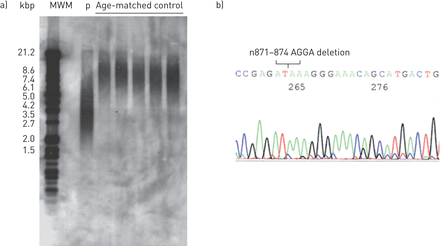
a)与年龄匹配的健康对照相比,Southern印迹分析显示患者(P)的较短端粒长度。MWM:分子量标记。b)通过直接测序的基因突变分析显示N871-874四核苷酸Agga缺失TINF2.基因。
Dyskeratiosis Congenita是一种稀有的遗传异源障碍,其特征是皮肤高度沉着,指甲缺乏和粘膜的白斑。骨髓衰竭是常见的发现,并已经注意到恶性肿瘤的倾向。虽然表达障碍症的肺部表现令人难以置疑是罕见的,但是欧卡尔[1[报道,在多达10-15%的患者中可能会看到异常的肺部特征。
遗传上,渗透症同仁塔基塔是异质的,已经确定了三种形式:X键的隐性,常染色体显性和常染色体隐性。在本案中,患者的父亲患有同样的疾病;因此,我们怀疑该患者的不良症的形式是常染色体的显性。血清瘤病患者的常染色体显性形式是由端粒酶的核心组分中的杂合突变引起的,TERC.[4.那5.),塔特[6.那7.]以及在避难所端粒保护复合物的组件中,TINF2.[3.].在这个病人中,突变TINF2.,但不TERC.和塔特,经基因突变分析证实。以前有报道说DKC1中[8.],TERC.[5.),塔特[6.]与Dyskeratosis Congenita患者的肺纤维化有关。DKC1中没有在这个病人中进行分析,因为突变在DKC1中导致x连锁形式的先天性角化不良。关于肺纤维化与TINF2.妊娠病变的突变,同胞,Walne等等。[3.据报道,只有一名患者在33例患有33例患者中患有其他临床特征的肺纤维化TINF2.突变。然而,他们没有详细描述患者。据我们所知,这是第一种表现出障碍症患者肺纤维化的案例报告TINF2.突变。
TINF2.据报道,突变是第六次发现的缺陷症Congenita基因中的杂合酶突变航空公司等等。[9.2008年)。TINF2.编码Tin2,是避难所端粒保护复合物的组成部分。避难所复合物对端粒至少有三种影响:它决定了端粒末端的结构,涉及通过端粒酶进行T圈的产生并控制整粒DNA的合成[1那10.].没有了庇护所的保护作用,染色体端粒不再隐藏在DNA修复机制中,染色体末端因此被DNA修复途径错误地处理。据报道,大约11%的先天性角化不良症是由TINF2.这些突变的突变和患者的端粒明显较短,而不是其他不良病变的同性恋亚型[3.].还据报道,大多数患有障碍症的患者TINF2.突变具有严重的疾病,而且与其他不良病变的同性恋基因相比,患者TINF2.突变在10岁以下之前具有增生性贫血的高发病率[3.].
肺泡上皮细胞凋亡增强的异常修复过程在肺纤维化如特发性肺纤维化的发病机制中起着至关重要的作用,但确切的机制尚不清楚。先天性角化不良肺纤维化的机制也尚未阐明。然而,由于先天性角化不良基因突变导致端粒长度短,端粒维持功能缺陷,肺泡上皮细胞的端粒可能较短。在先天性角化不良患者中,我们推测细胞死亡增强的异常肺修复导致肺纤维化,尽管肺泡上皮细胞的端粒短尚未被直接证实。
在此,我们描述了患有肺纤维化的疾病病变症的第一种病例报告TINF2.突变。本报告证明不仅是突变TERC.那塔特和DKC1中, 但是也TINF2.,导致疾病患者肺纤维化。但是,我们不知道为什么突变TERC.那塔特和DKC1中经常在腹膜炎症患者中发现肺纤维化患者与其他五个基因相反。此外,可以增加端粒酶活性的性激素是潜在的治疗药物;然而,没有在Dyskeratosis Congenita患者中确定用于肺纤维化的标准治疗。由于疾病疾病的肺纤维化的临床特征和发病机制,同性恋症患者不明确,案件的报告积累揭示了对这种破坏性疾病的理解。
脚注
利益冲突:没有宣布。
- 已收到2013年8月27日。
- 公认2013年9月4日。
- ©2013年











{kind=link}
{kind=link}