





摘要
肺泡上皮II型细胞是成人肺血管紧张素转换酶(ACE)-2的主要来源,通常是静止的,但在肺纤维化中活跃增殖并下调这种保护酶。因此,我们推测ACE-2的表达可能与细胞周期进展有关。
为了验证这一假设,我们检测了纤维化的人类肺中、肺泡上皮细胞系A549和MLE-12中融合后(静止期)的ACE-2 mRNA水平、蛋白水平和酶活性。与亚流畅(增殖)密度。
ACE-2mrna、免疫反应蛋白和酶活性在静止细胞中均较高,但在活跃增殖细胞中严重下调或缺失。转录阻断剂放线菌素D或c-Jun N末端激酶(JNK)抑制剂SP600125完全抑制了向静止状态发展的细胞中酶的上调。在特发性肺纤维化患者的肺活检标本中,肺泡上皮细胞中缺乏免疫反应酶,其增殖标志物阳性,但在缺乏增殖标志物的肺泡上皮细胞中大量表达。
这些数据说明ACE-2的肺纤维化的损失和由JNK介导的转录机制表明此保护酶的细胞周期依赖性调节。
介绍
大量调查的支持论点,即局部组织血管紧张素(ANG)系统在肺纤维化动物模型[发病的关键1,2]在特发性肺纤维化(IPF)中[3,4],由肺医师所遇到的最常见的和隐袭的间质性肺病(ILD)。在细胞和分子事件的信令为血管紧张素Ⅱ中起关键作用的证据点几行被认为是在肺纤维化的发病,包括肺泡上皮细胞(AEC)凋亡关键[五],成纤维细胞增殖和迁移[6,7]和胶原合成[8]。
细胞凋亡的培养的肺泡上皮的响应感应到各种促细胞凋亡和促纤维化刺激的[9-12]已显示既激活和需要血管紧张素原(AGT)的合成并且将处理后的肽血管紧张素Ⅱ,此系统的效应的肽。血管紧张素Ⅱ既是促运动[7和有丝分裂的[6]对人类肺成纤维细胞,增加胶原蛋白合成通过自分泌的机制介导的转化生长因子(TGF) -β1 transactivation纤维母细胞本身(8]。反过来刺激TGF-β1激活前胶原合成和肌成纤维细胞转变[13,14],除了在明显的自分泌循环中激活AGT表达[13]. ANG受体AT1阻滞剂对小鼠和大鼠肺纤维化的抑制作用[1,15]或AT1受体缺失小鼠[1]支持论点,即刚刚讨论的机制是活性体内以及在体外它们被首次鉴定系统。
我们实验室的证据支持血管紧张素转换酶(ACE)-2及其产物ANG1-7和ANG1-7受体mas组成的反调节轴在肺纤维化中的重要作用[五]。在小鼠或大鼠的肺纤维化的博来霉素模型,ACE-2被证明是通过干扰RNA(siRNA)使用小敲低或ACE-2的竞争性抑制与肽DX600,其中任一个加剧胶原沉积在保护响应于博来霉素[4]。其他作者也显示了ACE-2在单氟他林诱导的肺纤维化中的类似保护作用[16]。与此相反,施用纯化的重组ACE-2抑制博来霉素诱导的胶原沉积[4]。
在最近的一项凋亡培养肺泡上皮的调节的研究中,ACE-2和其产品的ANG1-7发现通过作用的ANG1-7的降低的c-Jun N-末端激酶(JNK)磷酸化的能力,以防止AEC死亡,对AEC的信令所需的事件凋亡响应于血管紧张素Ⅱ和其它促凋亡诱导剂[五]。的ANG1-7对AEC死亡的抑制作用是由的ANG1-7受体介导的MAS。这些结果与那些显示在前面的段落中所讨论的胶原沉积的抑制一起,证明ACE-2 /作用的ANG1-7 / MAS轴线在实验性肺纤维化的抗凋亡和抗纤维化作用。
我在研究中的一个重要发现一世等。[4]是在实验和人类肺纤维化中保护酶ase -2均下调的证据。IPF患者活检获得的人肺组织中,ACE-2在mRNA、免疫反应蛋白和酶活性水平上均降低,且均降低到相似的严重程度。同样,在博莱霉素诱导的小鼠和大鼠IPF模型中,ACE-2蛋白、酶活性和mRNA也降低了,但在暴露于博莱霉素的培养AECs中,ACE-2 mRNA未见降低体外。出于这个原因,我们试图找到可以解释的是从来没有接触到bleomyin或凋亡的其他外源性诱导纤维化人体肺部ACE-2基因,蛋白和活性丧失的机制。
多年纤维化人肺的肺泡上皮细胞已被描述为“增生”或“立方”上皮,基于所述观测主要II型肺被增殖响应于正在进行的上皮损伤[17,18]。与此相反,正常肺的肺泡上皮细胞基本上是静止的,具有很少或没有增殖细胞和大量的I型细胞,类型的终末分化的子代II细胞[19]。在此基础上,人们假设,在ACE-2的减少纤维化人体肺部[观察4]可以是由II型肺细胞周期进展的结果。我们在这里报告,ACE-2基因,免疫活性蛋白和酶活性都有很高的肺泡上皮中是休眠表达,而在已经进入细胞周期肺泡上皮被下调的发现。我们还报告的证据表明,ACE-2的伴随肺泡上皮对静止进展的上调是转录由JNK介导的机制调节。
材料和方法
物料
荧光肽底物Mca-YVADAPK(DNP)-OH和人重组ACE-2购自R&d系统(明尼阿波利斯,MN,USA)获得纯化。肽DX600是由大分子结构基金(生物化学,密歇根州立大学,东兰辛,MI,USA的部门)合成。针对ACE-2和抗体anticytokeratin MNF-116抗体购自Abcam公司(剑桥,MA,USA)获得。抗增殖细胞核抗原抗体(PCNA)和溴脱氧核苷(BrdU)来自BD Biosciences(圣地亚哥,CA,USA)购得。放线菌素d和SP600125购自Sigma-Aldrich公司(圣路易斯,MO,USA)购买。SB203580从细胞信号技术公司(丹弗斯,MA,USA)获得,PD98059购自Invitrogen(格兰德岛,NY,USA)购买。对于ACE-2的逆转录酶PCR(RT-PCR)引物,来自Santa Cruz生物技术公司(Santa Cruz公司,CA,USA)购买。所有其它材料均是试剂级的。
细胞培养
人肺腺癌细胞系A549来自美国型细胞培养收集,在含10%胎牛血清(FBS)的Ham F12培养基中培养。小鼠肺上皮细胞系MLE-12是J. Whitsett (University of Cincinnati, Cincinnati, OH, USA)实验室赠送的礼物,在添加了5%胎牛血清的完整HITES培养基中培养。所有实验均在有血清的情况下进行,而不考虑细胞密度。在60-75%的融合处收集次融合细胞,在融合后5天收集后融合细胞,除非另有说明。
人体组织样本和处理
通过在胸部诊所的方向,Bellvitge大学附属医院,L'Hospitalet的Llobregat的,西班牙进行电视胸腔镜肺手术获得人肺组织。从14名IPF患者获得纤维化肺组织;来自多于一个肺叶得到活检。所有患者均是符合诊断标准的IPF [临床,功能性,放射性和组织学特征20]。两位不同的放射学专家和病理学家分别对高分辨率x线断层扫描图像和肺切片的组织学模式进行了评估。患者既无任何职业性或环境接触史,也无其他已知的固体废物成因。ILDs多学科委员会对所有病例进行了评估。这些IPF患者在采集肺样本时均未接受类固醇或其他免疫抑制剂治疗。7例无肺部疾病史的自发性气胸患者接受手术治疗,获得正常的肺组织。在这些组织样本中没有发现疾病的组织病理学证据。根据机构指南,获得患者的书面知情同意,该研究获得了贝尔维奇大学医院伦理委员会的批准。所有组织固定于10%中性福尔马林缓冲液中16 h,石蜡包埋。部分被削减为4.0μm厚度和安装在玻璃盖玻片。
RNA分离和RT-PCR
总RNA从根据制造商的说明书的人肺活检或用Trizol试剂(Invitrogen)冷冻小鼠肺提取。第一链cDNA用2-微克用Superscript II逆转录酶(Invitrogen)和寡聚(dT)12-18的总RNA合成。实时RT-PCR进行的cDNA从50合成总RNA纳克,根据制造商的方案,和0.2人类μM特异性引物的SYBR Green PCR核心试剂(Applied Biosystems公司,福斯特城,CA,USA)ACE-2(有义5'-CATTGGAGCAAGTGTTGGATCTT-3' 和反义5'-GAGCTAATGCATGCCATTCTCA-3 ')和β-actin(有义5'-AGGCCAACCGCGAGAAGATGACC-3' 和反义5'-GAAGTCCAGGGCGACGTAGC-3' )。对于小鼠ACE-2,引物为:有义:5'-GGATACCTACCCTTCCTACATCAGC-3' 和反义5'-CTACCC CACATATCACCAAGCA-3' 。对于小鼠β肌动蛋白,引物为:有义:5'-TCCTGTGGCATCCATGAAACT-3' 和反义5'-CTTCGTGAACGCCACGTGCTA-3' 。开始用10分钟的激活的Taq的PCR热循环模式聚合酶在95℃,然后变性的40个循环的94℃持续60秒,在55退火℃60秒,72℃60秒延伸,结束解离曲线分析,以验证PCR产物的特异性。反应在Mx3000P进行机(Stratagene公司,拉霍亚,CA,USA)和循环阈值(执行ÇŤ)数据用的MxPro-Mx3000P进行软件版本3.0(Stratagene公司)收集。相对ACE-2的表达归一化至β肌动蛋白,并与比较例C计算Ť的2方法−ΔΔCt。在所有附图中,对于ACE-2的平均值/β肌动蛋白对照组比设定为100%,并且相对于ACE-2 /β肌动蛋白之比为治疗组中表达。
Western印迹
用冰镇np40为基础的裂解缓冲液对人或小鼠肺上皮细胞进行均质化,加入蛋白酶抑制剂(蛋白酶抑制剂鸡尾酒P840;Sigma-Aldrich)。可溶性蛋白提取物(20μg) Tris-HCl加载并运行在10%聚丙烯酰胺凝胶,通过SDS - page分离,在10×三羟甲基氨基甲烷/甘氨酸/ SDS缓冲液(Bio-Rad、大力神、钙、美国)。凝胶转移到免疫印迹PVDF印迹膜(Bio-Rad)的托宾缓冲液。在0.1%吐温20的三倍缓冲盐水中,用5%脱脂奶粉堵塞吸膜。采用抗ACE-2多克隆抗体(1:200稀释;圣克鲁斯生物技术公司)。用辣根过氧化物酶(HRP)标记的驴抗山羊二抗(1:2000稀释;圣克鲁斯生物技术公司)使用化学发光底物超信号西飞到最高灵敏度(皮尔斯,罗克福德,伊利诺伊州,美国)。确保相同加载的蛋白,膜被然后reprobed抗体对β-actin(细胞信号技术,Inc .)。
ACE-2酶测定法
蛋白从人活检样品或从小鼠肺进行匀浆在冰冷的Tris-HCl无EDTA缓冲液提取,pH为6.5 [4]。ACE-2蛋白的酶活性,在10μM均质化通过荧光底物MCA-YVADAPK的切割后立即测量,在肺组织匀浆45μL用Tris-HCl缓冲液pH为6.5的含赖诺普利(50微克·L-1)阻断ACE活性[4,五]。反应在室温下的黑色微滴定板上进行,使用荧光微平板阅读器(FL600 Biotec荧光阅读器;超过30分钟,使用310/320和420/450 nm的激发和发射波长。
免疫组织化学和免疫细胞化学
免疫组化检测ACE-2、II型细胞特异性细胞角蛋白和PCNA,使用抗ACE-2抗体(Abcam;1:50稀释),抗细胞角蛋白抗体MNF-116 (Abcam;1:50稀释)和抗pcna单克隆抗体(BD生物科学;1:10 0稀释)。用含3%牛血清白蛋白的PBS溶液封闭肺切片1小时;在3%牛血清白蛋白/PBS中,在4℃下过夜。PBS洗涤后,用生物素偶联的二抗和亲和素连接的色原系统检测抗体。致色原要么是二氨基联苯胺(棕色),要么是坚牢红(红色)。如前所述,在固定乙醇的AEC单分子层上进行免疫组化[21],使用荧光素异硫氰酸酯(FITC)结合的抗brdu抗体(BD生物科学)或用于western blotting的相同的ACE-2抗体。
显微镜,图像采集和流式细胞术
将制备的肺切片下发送或落射荧光光拍摄上装有一个滑块SPOT数码相机(SPOT成像解决方案,斯特林高地,MI,USA)的Olympus BH2落射荧光显微镜(奥林巴斯公司,东京,日本)。绿色荧光(抗ACE-2或BrdU的)的图象通过一个520纳米的带通滤波器获得的。二元流式细胞术数据结合的BrdU与如先前所述进行DNA分布[22]对细胞通过胰蛋白酶消化收获细胞。流式细胞术数据被收购上的Accuri C6激光流式细胞仪(BD Accuri流式细胞仪,安阿伯,MI,USA)。
结果
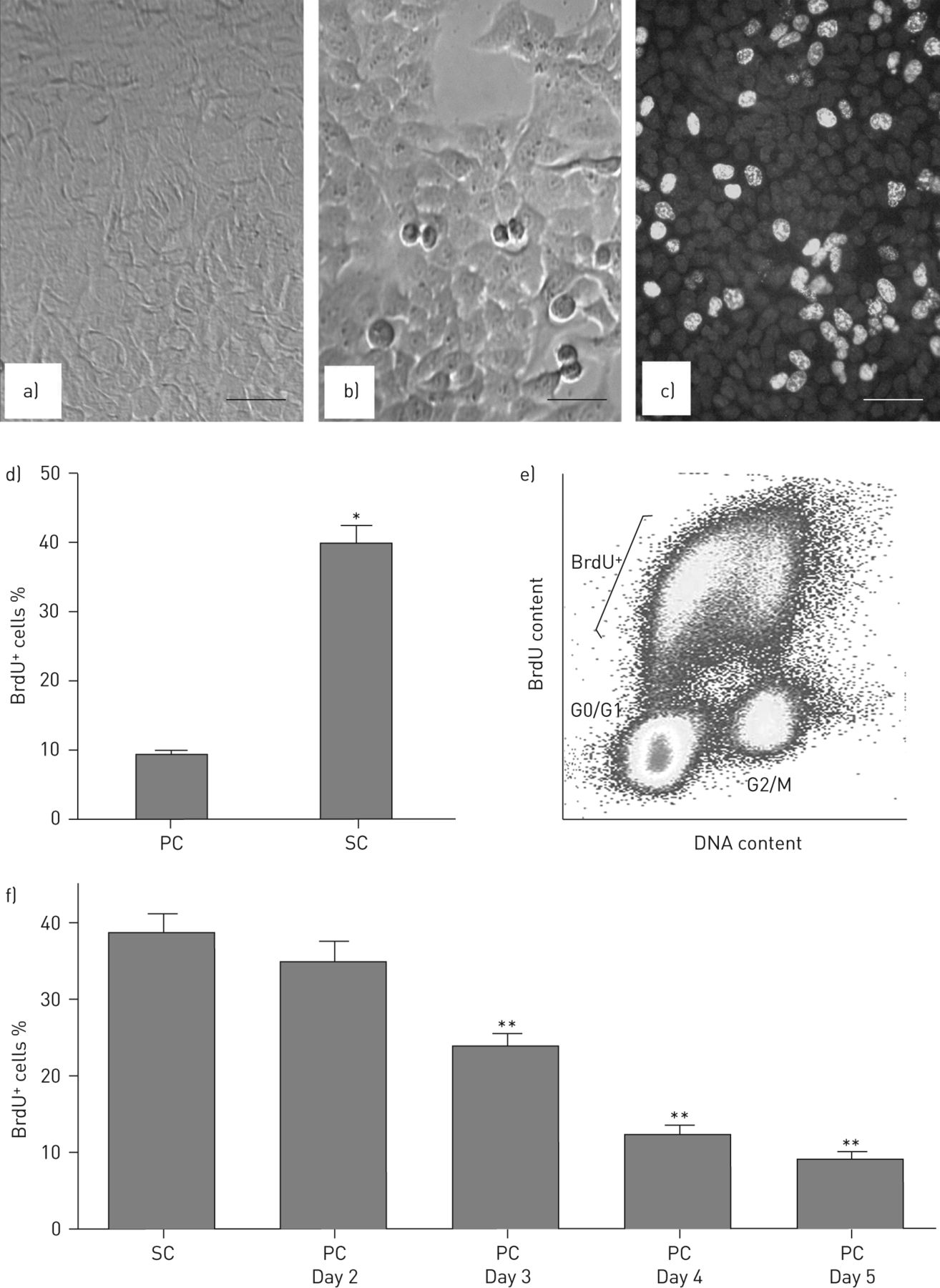
由于上述原因,ACE-2蛋白水平,酶活性和mRNA在人进行了检查,鼠标AEC系A549和MLE-12下增殖与静态培养条件下,无论是在生长因子的存在。图1a和b融合后和亚汇合培养条件下培养显示了人肺泡上皮的A549细胞,分别,无论是在生长因子的存在(即无论是在血清的存在下)。图1c示出的BrdU S期细胞的标记,其在定量图1D并汇合的培养条件下显著上升。通过荧光激活细胞分选仪分析(图。1E), brdu阳性细胞在早期、中期或晚期的均匀分布表明,在亚融合培养条件下,brdu阳性细胞确实在进行复制DNA合成[21]。图1F演示了BrdU阳性S期细胞伴随从对数期生长中汇合培养到静止的进展在融合后的第5天类似的结果在培养的小鼠中观察到AEC MLE-12(数据未示出)的下降。
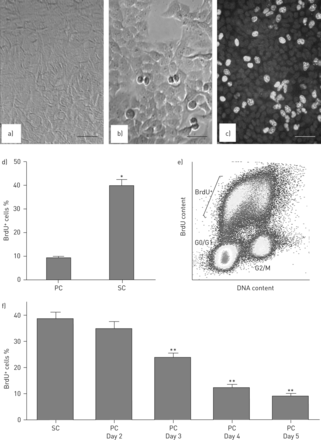
细胞周期在人肺上皮细胞中的调控作用。如材料和方法所述,在亚融合(SC)或后融合(PC)条件下培养人肺泡上皮细胞系A549,均存在生长因子。a) PC第2天A549细胞的相衬显微照片;b) SC A549细胞的相衬显微照片;注意:双核细胞胞质分裂。c) SC培养条件下s期A549细胞荧光素异硫氰酸酯(FITC)溴脱氧尿苷(BrdU)标记。d) PC培养条件下brdu阳性细胞核百分比与SC。数据表示为平均值±SEM至少有三种细胞培养。*:p<0.05与电脑通过t检验。e) DNA分布的双变量流式细胞仪分析(x轴)与SC A549细胞的结合的BrdU(y轴);注意BrdU阳性细胞的跨S期DNA含量的均匀分布[21]。f) A549细胞从次融合到融合后第5天BrdU标记降低的定量。数据表示为平均值±SEM至少有三种细胞培养。比例尺=50μm**:p<0.01与通过方差分析和Newman-Keul检验。详情见正文。
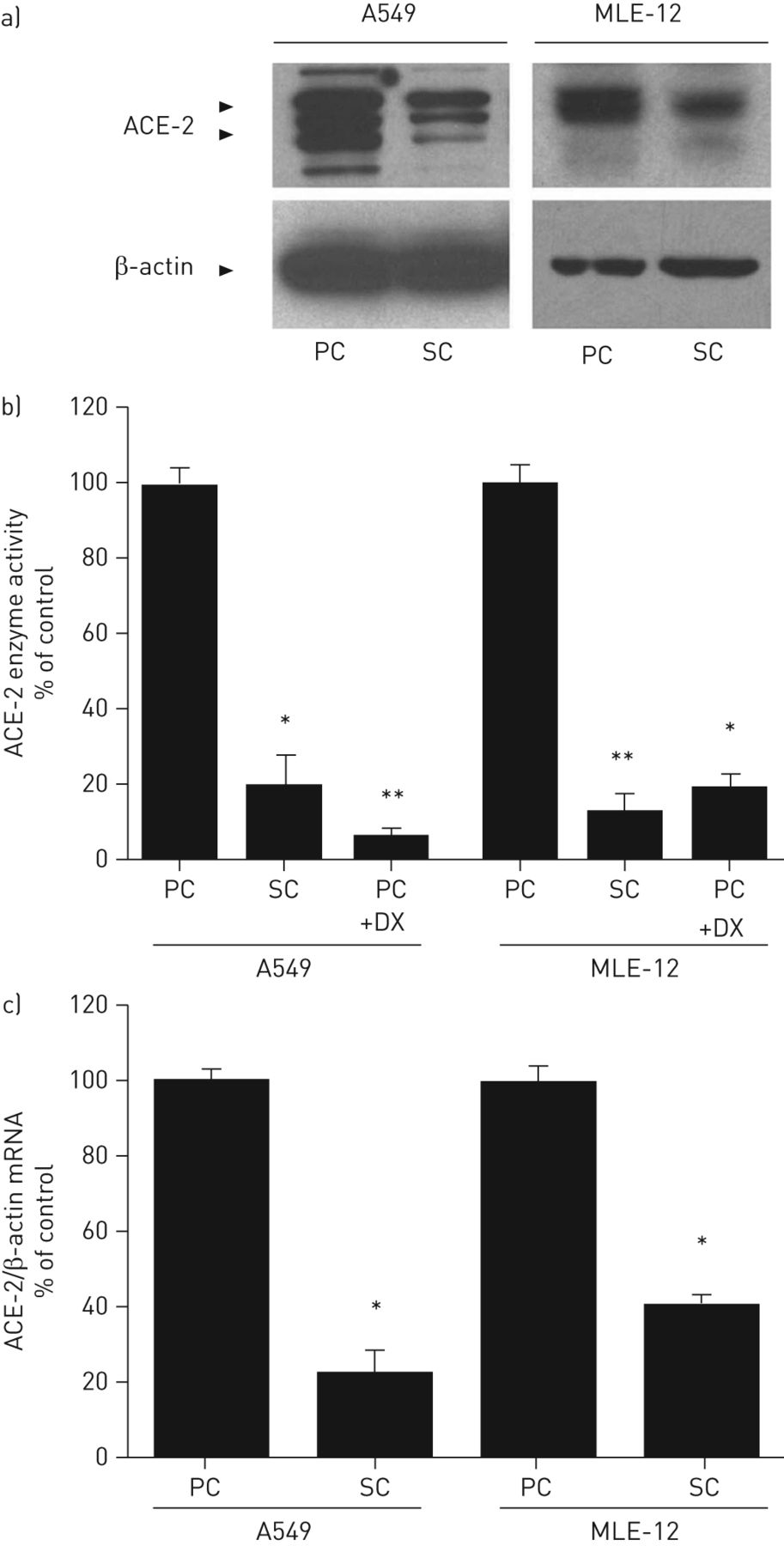
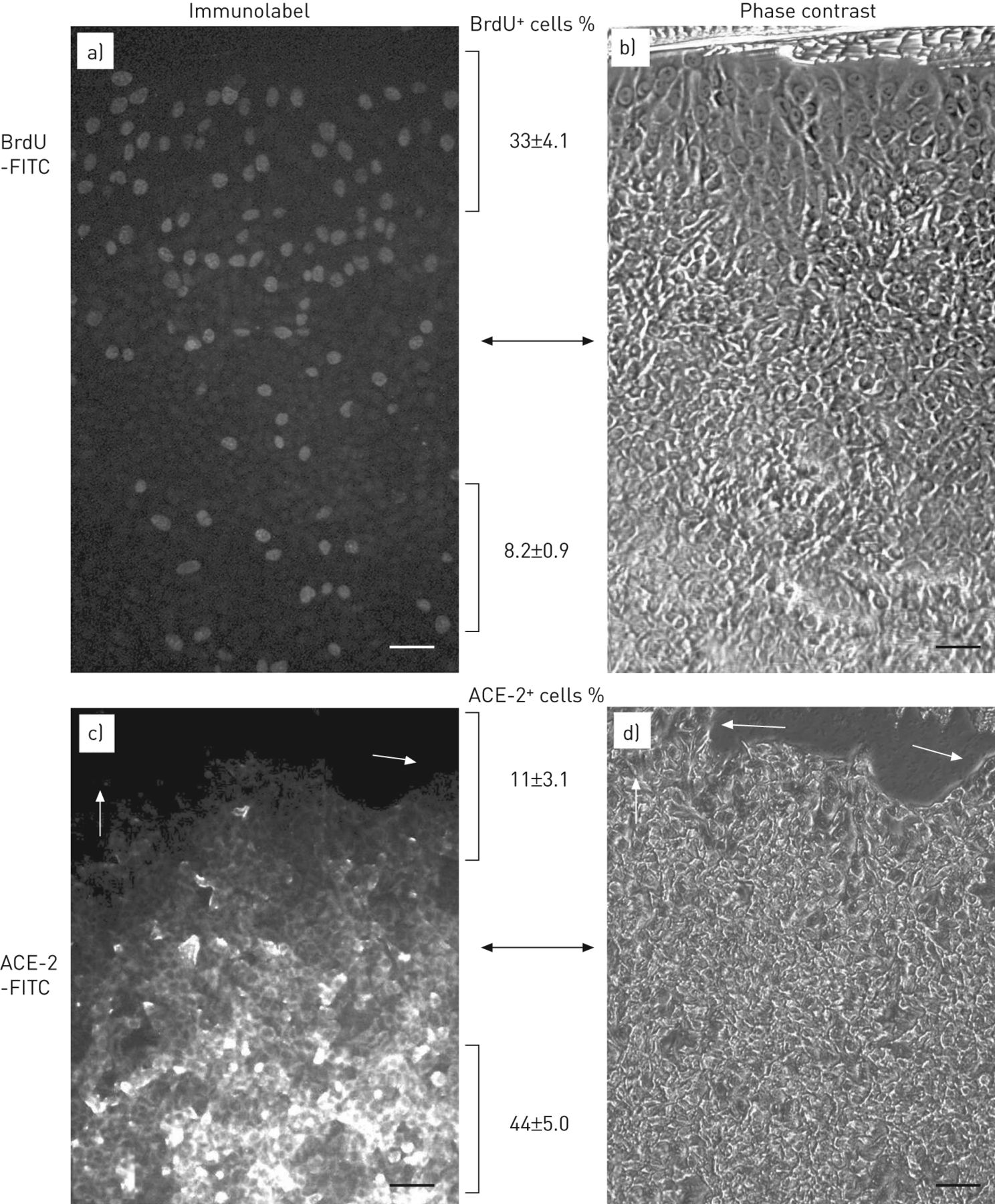
图2示出了削减ACE-2免疫反应性蛋白(图2a),酶活性(图2b)和ac -2 mRNA (图2c)在相对于静止(融合后第5天)的条件下,无论是在生长因子的存在下增殖(亚汇合培养的人A549细胞和小鼠MLE-12细胞)。对于各细胞类型,酶的活性通过荧光肽底物检测(图2b;通过添加竞争性的ACE-2抑制剂肽DX600 [23]。如在此描述的进行,当这些数据证实所述活性测定的特异性。在图2c,mRNA表达的ACE-2降低到类似于用于ACE-2免疫反应性蛋白或酶活性中观察到的程度。在一个不同的实验方法,将A549细胞培养至融合后静止的条件(在血清的存在下)进行的实验伤人(该体外“从头开始”模型)来诱导紧邻划伤细胞增殖。图3显示融合后对BrdU免疫的A549细胞(图。图3a和b)或ACE-2(图3 c与划痕相邻的区域(每个面板的顶部)以及更远的区域(每个面板的底部)。在图3a,将细胞以划痕的边缘显示增加BrdU标记指数(BrdU的+相对于细胞的细胞%)远离划伤。图3c与d显示沿划痕边缘增殖细胞的ACE-2免疫反应活性降低(白色箭头,图3 c),在同一微观场的相衬图像中清晰可见(图3 d黑色的箭头)。
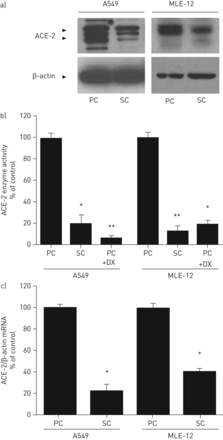
血管紧张素转换酶(ACE)-2蛋白,酶的活性和mRNA与人类和小鼠的肺泡增殖培养物中上皮细胞的下调。肺泡上皮细胞系,A549和MLE-12的人类和小鼠,分别在融合后(PC)和亚汇合(SC)的密度如在材料和方法中所述和随后收获用于一个)western印迹,B)ACE培养通过逆转录酶PCR -2酶测定或c)mRNA分析。一)的来自三个独立实验的三个相似印迹的代表性示例。b)中的裂解物从PC条件下培养的细胞也测定在肽DX600的存在(DX),ACE-2的竞争性抑制剂。注意由DX(1μM)的基本上完全的抑制。数据表示为平均值±SEM至少有三种细胞培养。*:p<0.05与PC;**:P <0.01与通过方差分析和Newman-Keul的测试PC。
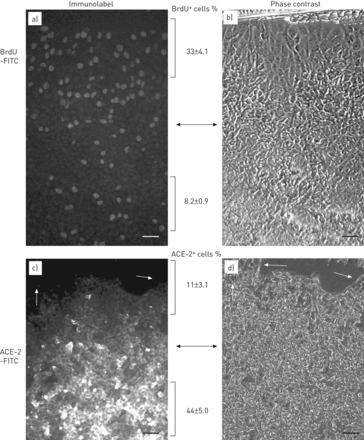
在人肺泡上皮细胞血管紧张素转换酶(ACE)-2免疫反应的下调修复伤口体外。人细胞系A549培养以融合后密度,并进行由划痕单层伤人后跟一个短时间暴露于溴脱氧尿苷(BrdU的)(见材料和方法)。然后单层固定,并用异硫氰酸荧光素(FITC)缀合的免疫标记的抗体抗BrdU的(a和b)或ACE-2(c和d)。免疫标记(a和c)和相位对比(b和d)相同的显微镜视野的图像被示出。总之,划痕是在顶部。注意紧邻其大多是阴性的ACE-2免疫反应性(C,白色箭头)的scatch(d,黑色箭头)的细胞的区域。还要注意紧邻当与密度远离刮(一,下腹)相比,暂存(一个,上部象限)BrdU阳性核的密度增加。BrdU阳性核和ACE-2阳性细胞的定量在划痕的边缘与在单层的中心表示。数值是均值±SEM至少三个细胞培养物;既BrdU阳性和ACE-2阳性值是由t检验P <0.05显著。比例尺=50μm以下。
以确定是否可以检测到ACE-2与上皮细胞增殖之间的这种反向关系体内响应于肺泡上皮的生长因子诱导的细胞增殖,C57BL6小鼠用角质形成细胞生长因子(KGF)气管内滴注,已知的上皮特异性有丝分裂原活性的肺泡上皮[24,25]。图4示出了用于免疫标记ACE-2的(a,c和e)和BrdU(b和d),每个在相邻的连续切片进行,以确保在同一微环境内共定位。在正常小鼠(SHAM-灌输),ACE-2免疫标记是在肺泡角内上皮细胞健壮(图4,棕色),但同一微环境中的BrdU免疫标记很少(图4 b). 相比之下,在气管内滴注后2天检查的KGF滴注小鼠显示出减少的ACE-2免疫标记(图4摄氏度)在相同的微环境中,BrdU标记是稳健的(图4 d)。图4 e显示KGF诱导的AEC增殖在KGF灌注2周后恢复了强健的aca -2免疫水平,这段时间KGF诱导的AEC增殖已经消退[24]。图4 f证明在KGF灌注后2天,肺组织总ACE-2酶活性显著降低。
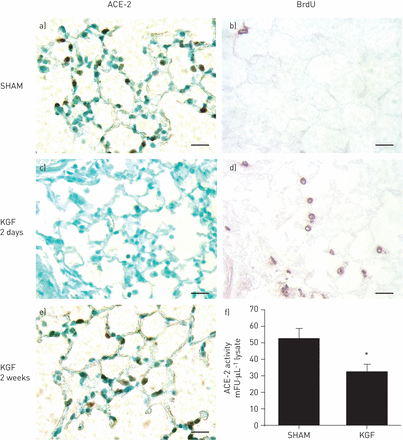
在增殖与角质细胞生长因子滴注小鼠肺上皮细胞的(KGF)血管紧张素转换酶(ACE)-2的下调原位。用纯化的重组KGF或假性载体(vehicle, SHAM)灌胃C57BL6小鼠。献祭前1小时,动物给予溴脱氧尿苷(BrdU;50 mg·公斤-1腹膜内用于增殖细胞[鉴定。)21]。肺切片与抗体免疫标记针对ACE-2(a,c和e)或BrdU的(b和d)。F)将肺均化ACE-2酶测定。数据表示为平均值±SEM的n = 4的每一个用于SHAM和KGF天2.比例尺= 50微米。傅:荧光单位。*:P <0.05。见材料和方法的详细信息。
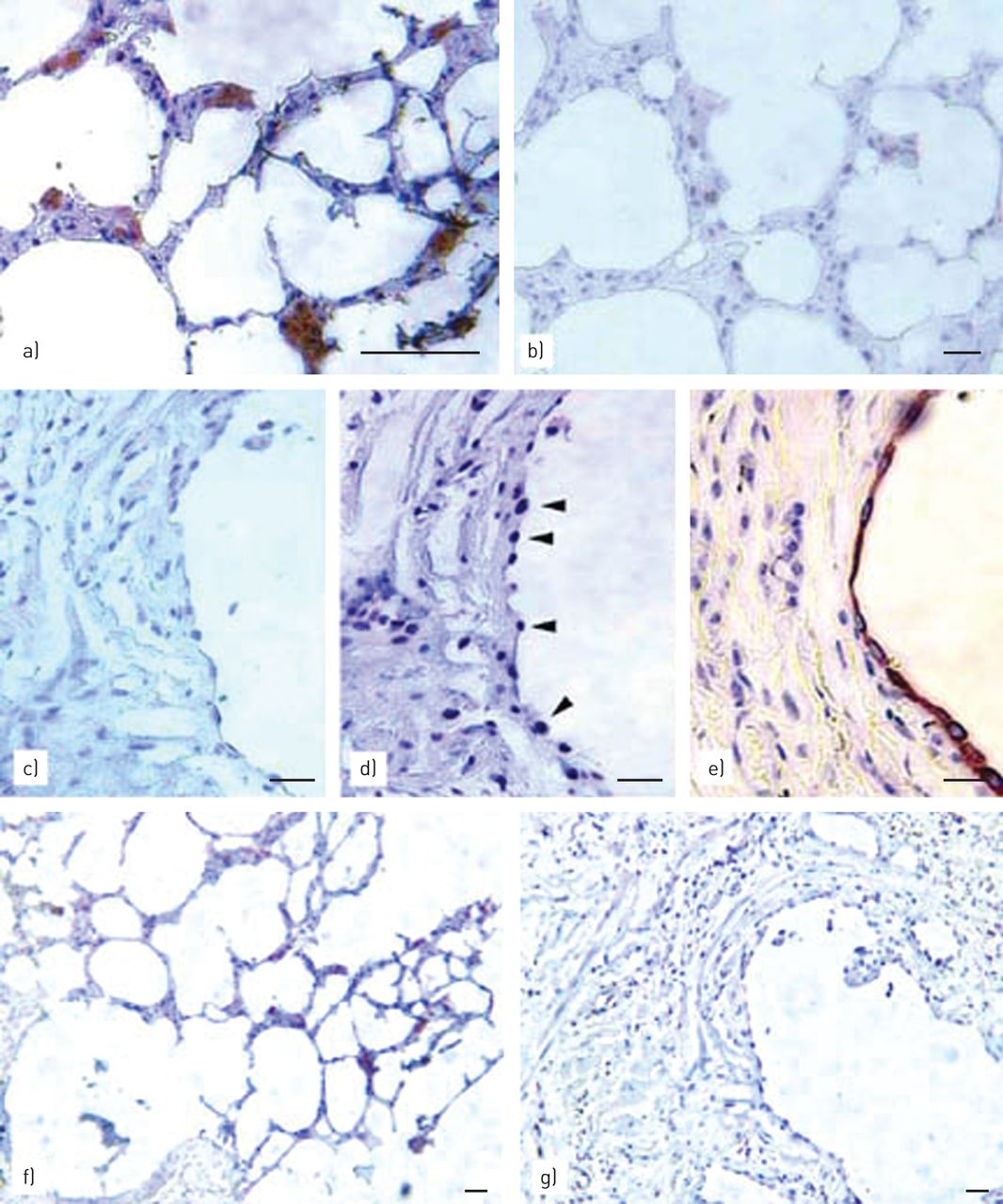
要确定是否ACE-2和上皮细胞增殖之间的这种反比关系可能会在完整的人肺被检测到,从IPF患者或正常人的肺获得活检标本进行免疫组织化学对于ACE-2,PCNA和的特异性免疫标记肺泡上皮(单克隆抗体MNF-116 [24])。图5显示,在正常人肺,免疫反应性ACE-2很容易在肺泡角内上皮细胞中检测到(图5a,棕),II型肺的正常位置。免疫组织化学对PCNA在相同的微环境的串行部分执行显示没有增殖的上皮细胞(图5b),具有式的记录静止II肺在正常,未受损伤的肺[一致19,24]。相比之下,对来自IPF患者的肺活检(图5c)基本上揭示了一个微环境的肺泡上皮,其中上皮被严重标记与PCNA没有ACE-2免疫反应性(图5d,箭头),如在相邻的连续切片进行检测。图5 e免疫组织化学描绘用于单克隆抗体MNF-116(棕色)在第三相邻连续切片,以揭示上皮层确实由ACE-2和PCNA抗体二者访问。ACE-2阴性的支气管上皮细胞也被在IPF肺组织中观察到(未示出)。
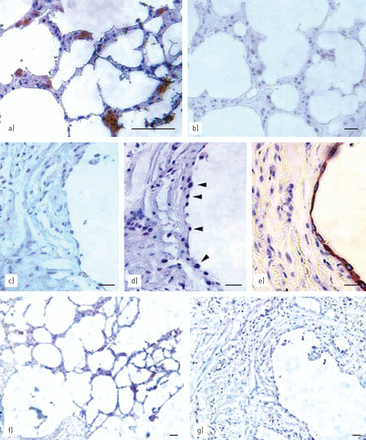
在增殖人类特发性肺纤维化的上皮血管紧张素转换酶(ACE)-2的下调(IPF)肺原位. 对正常人肺(a、b)和IPF人肺(c、d、e)的石蜡切片进行ACE-2、增殖细胞核抗原(PCNA)或MNF-116的免疫组化染色。a) 正常人肺泡角细胞ACE-2(棕色)的重标记。b) PCNA的阴性标记(与d相反)在同一微环境中的连续切片中,与a中分析的切片相邻。c) IPF人肺上皮细胞ACE-2的阴性标记(与a对照)。d) 在IPF肺的同一上皮(与c相邻的一系列切片)中进行PCNA(箭头)重标记。e) 抗体MNF-116(棕色)在另一个连续切片(与d相邻)上的阳性标记识别了c)和d中研究的上皮层。f) a)来源于同一区域的低倍视野;棕色:ACE-2阳性细胞。g) 与c)相同区域的低倍视野。有关患者群体的详细信息和描述,请参见材料和方法。比例尺=50μm。
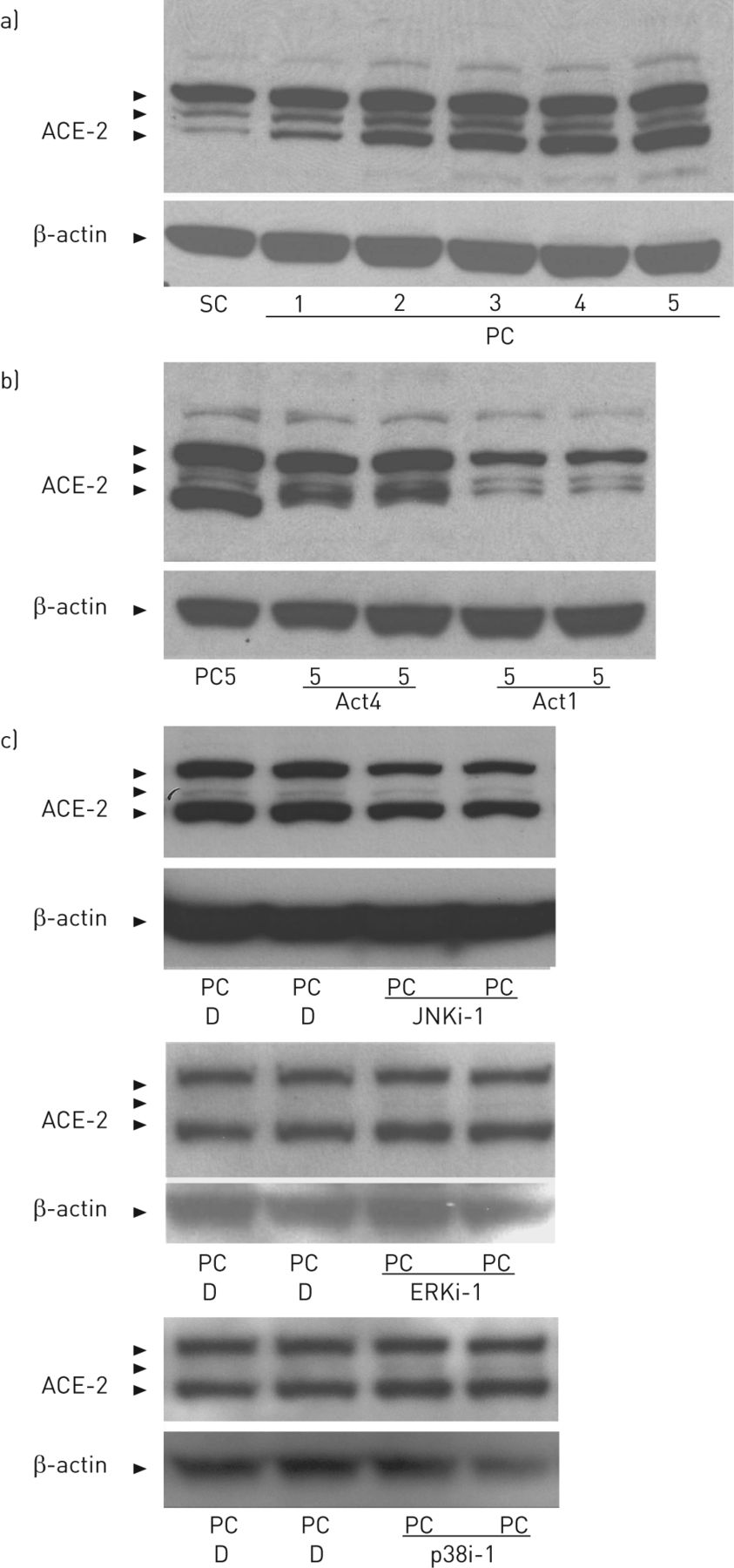
在图6,文化系统的次融合与融合后的人肺泡上皮用于开始确定可能从汇合调节ACE-2的表达作为肺泡上皮转变的机制,增殖细胞到静止的,融合后的细胞。图6显示,在亚汇合的人A549细胞增殖的培养物(以相对少的ACE-2)逐渐积累多个免疫ACE-2的每一天在融合后的培养。在融合后的培养物,细胞增殖增加的时间(由BrdU标记评估)逐渐降低至在所示的5天的融合后值图1b(未显示BrdU时间进程)。图6 b示出了转录抑制剂放线菌素d,如果添加到培养基中在任postconfluence天4或postconfluence 1天,可以部分或完全阻断,分别免疫反应ACE-2蛋白的积累,将通过第5天postconfluence否则发生。图6 c显示,SP600125,JNK的抑制剂,可以抑制在A549细胞中的ACE-2的积累如果融合后培养添加到培养基上的第5天。与此相反,细胞外信号调节激酶(ERK)或P38介导的信号的抑制剂对ACE-2的积累没有抑制作用。这些数据表明,发生作为肺泡上皮过渡从增殖到静止期培养物在ACE-2的增加是由JNK介导的转录的机制调节。

证据的c-Jun N-末端激酶(JNK) - 介导血管紧张素转换酶(ACE)-2上调的转录控制在从电池循环到静止的上皮转化。人细胞系A549在亚汇合密度培养,并过渡到融合后静止期间每天监测通过western印迹,无论是在放线菌素d(ACT),载体(0.1%DMSO在F12培养基; d)存在或不存在的或抑制剂JNK(JNKi)的ERK(ERKi)或P38(p38i)介导的信号传导途径。见材料和细节的方法;所有面板显示来自三个独立实验的至少三个相似印迹的一个代表性的例子。一)在免疫反应性ACE-2增加作为时间(天)在融合后(PC)培养相对于亚汇合(SC)培养物的功能。B)ACE-2的上调超过5天的A549细胞的PC培养(PC5)的阻断通过加入放线菌素d(1微克·毫升-1)第4天(Act4)或第1天(Act1)。注意ACE-2表达式的相似性Act1条件下观察SC文化(在)。c)堵塞ACE-2 upregulation超过5天的电脑文化的A549细胞的SP600125 postconfluence天(10μM) 1 (JNKi-1),但不是通过erk抑制剂(PD98059 10μM)或p38 (SB203580 10μM)应用于相同的方式。
讨论
据我们所知,这是ACE-2的表达调控细胞周期的任何细胞类型或器官的功能的第一份报告。一般地,ACE-2的表达的控制是知之甚少。在心肌细胞和成纤维细胞,ACE-2的mRNA和蛋白是由血管紧张素Ⅱ或内皮素(ET)-1下调和上调由所述ANG受体1阻断剂氯沙坦[26]。ANGII或ET-1的作用被丝裂原激活蛋白激酶-1的抑制剂所抑制,提示这些肽的作用可能是通过ERKs介导的。ANGII的ACE-2降解产物之一ANG1-7也通过ANG1-7受体mas抑制ANGII或ET-1引起的ACE-2 mRNA的减少。
在小鼠肺,ACE-2已经显示出在18.5天胚胎被发育调节,mRNA的最高的,并且要在细支气管和肺泡上皮细胞[主要表达27]。在人肺,气道上皮细胞是通过肺部感染期间SARS冠状病毒的第一个接触点中的一个;此外,ACE-2已被证明是该网站到SARS病毒结合以引发组织感染[28]。在培养的人类气道上皮细胞的研究中,脱落ACE-2胞外结构域的被认为是SARS感染的程度和预后的重要决定因素[29]。有关体外研究已显示,在ACE-2胞外域脱落是通过佛波醇酯上调,而且,依赖于钙调蛋白的一个特定的在ACE-2的胞质尾结合结构域的结合三十]。虽然脱落的ACE-2被认为是重要的SARS感染,这些机制的决定因素之一的ACE-2体外域脱落,如果有的话,对肺部ACE-2的生理作用的影响,目前还不清楚。
在暴露于缺氧条件下培养人肺动脉平滑肌细胞体外,ACE-2的mRNA和蛋白瞬时表达上调的方式依赖于转录因子低氧诱导因子1α〔31]。然而,ACE-2,在这些细胞中由血管紧张素Ⅱ由AT1受体的拮抗剂而不是由AT 2受体拮抗剂下调的方式可抑制。除了这两项研究,知之甚少调节在肺细胞ACE-2基因表达的因素。虽然作品上述表明,在肺泡上皮ACE-2水平可能是响应于血管紧张素Ⅱ或的ANG1-7的稳态水平的变化(即的反馈回路)的细胞增殖的独立的,在加入ACE-2抑制剂DX600的[五]在从亚流式培养到5天后流式培养的过程中(如图6)对ACE-2的积累没有明显的影响(数据未显示)。
本文报道的数据有力地支持了ACE-2在AECs中的表达受细胞周期依赖性调节的假设,因为在融合后的培养中,相对于静止的细胞,ACE-2 mRNA、蛋白和酶活性在增殖AECs时均降低。虽然fluorogenic肽底物使用,在其他地方,测量已知ACE-2也为ACE-1作为衬底,interleukin-1β-converting酶和其他肽酶(4],ACE-2活性通过添加竞争ACE-2抑制剂肽DX600的消除(图2)支持了我们的观点,即这里使用的产生特异于ACE-2测量ACE-2的测定条件。
类似地,在紧邻an的A549细胞中发现ACE-2免疫反应活性降低或缺失体外伤口 (图4)或具有稳健上皮细胞增殖的纤维化人肺的区域(内图5)反对细胞培养伪像中观察到增殖的贡献的实质差异ACE-2的表达与融合后的AEC文化。在此基础上,它是假设,增殖的细胞培养模型与融合后的AEC线,本文中所描述,提供了一个可行的实验系统,用以开始探索ACE-2的表达在细胞周期依赖的方式调节的分子机制。我们的背后ACE-2的上调作为培养的肺泡上皮进入静止的机制的初步研究(图6)是与工作假说,即ACE-2基因的转录是由细胞周期通过JNK介导的机制增加了出口相一致。尝试识别转录因子和JNK依赖性信号转导途径的活性在此过程中是目前正在进行中。
这种细胞周期依赖性的ACE-2调控机制的潜在生理意义目前尚不清楚,但在当前关于人肺纤维化肺泡上皮的思考背景下,这可能是合理的。纤维化肺中“增生”或“立方形”肺泡上皮的经典观察结果与长期以来的观点一致,即持续的上皮损伤刺激通过II型肺细胞增殖修复上皮损伤的尝试[18,19,32]。尽管肌成纤维细胞灶底层在IPF异常上皮被认为影响上皮细胞的存活[3,13,在这里研究的IPF肺活检中,ACE-2的下调似乎与肌成纤维细胞病灶的存在或不存在没有空间关系(数据未显示)。上皮细胞未能完全修复和替换正常的II型和I型细胞群,这曾被认为主要是增殖或分化失调的结果[33],在与AEC增殖相同的微环境中,上皮细胞凋亡的一致性观察使之复杂化[3,34,35]。
这两种意见,虽然看似矛盾,在有关细胞分裂和细胞死亡的信号电流知识之光认为是有意义的;在任一细胞增殖或细胞凋亡的控制,需要细胞周期进展两个初始信令和执行[36]. 此外,最近的研究表明血管紧张素系统,特别是ACE-2,其产物ANG1-7和JNK,在调节AECs凋亡反应中起着关键作用[五]。事实上,人类的肺肌成纤维细胞凋亡和肺泡上皮已被证明合成和分泌血管紧张素Ⅱ体外[9,10,13]并表示在纤维化人肺ANG肽[3]。鉴于这一发现ACE-2由血管紧张素Ⅱ的方式可抑制在心肌细胞和成纤维细胞[下调通过作用的ANG1-726],这将是非常令人感兴趣的研究,同样是这些ANG肽可能在人的肺纤维化肺泡上皮细胞发挥细胞周期依赖性在ACE-2基因表达的下调具有调节作用的可能性。在这方面,一些作者进行了偶然使用ACE抑制剂的回顾性分析IPF患者和报道没有益处[37]。然而,如通过乙最近讨论udinger[38],这些研究反对ANGII在人类肺纤维化中发挥促纤维化作用的结论未能确认ACE-2产物ANG1-7的抗纤维化作用[五,16],通过给药ACE抑制剂可使其减少。由于这些原因,血管紧张素受体阻滞剂,而不是血管紧张素转换酶抑制剂,目前在IPF患者的临床试验中备受关注[25]。
脚注
支持声明:该作品是由PHS HL-45136和支持美国心脏协会资助0950045G(授予B.D.乌哈尔),并由西班牙迪贝尔授予M. Molina-Molina。
利益冲突:无申报。
- 收到了2012年1月25日。
- 公认2012年9月30日。
- ©ERS 2013











![Manipulation of cell cycle status in cultured human lung epithelial cells. The human alveolar epithelial cell line A549 was cultured under subconfluent (SC) or postconfluent (PC) conditions, both in the presence of growth factors, as described in the Materials and Methods. a) Phase contrast micrograph of A549 cells at PC day 2; note the lack of binucleated or mitotic cells (compare to b). b) Phase contrast micrograph of SC A549 cells; note binucleated cells undergoing cytokinesis. c) Fluorescein isothiocyanate (FITC) bromodeoxyuridine (BrdU) labelling of S-phase A549 cells under SC culture conditions. d) Percentage of BrdU-positive nuclei under the culture conditions PC versus SC. Data are presented as the mean±sem of at least three cell cultures. *: p<0.05 versus PC by t-test. e) Bivariate flow cytometric analysis of DNA distribution (x-axis) versus incorporated BrdU (y-axis) of SC A549 cells; note the uniform distribution of BrdU-positive cells across S-phase DNA content [21]. f) Quantitation of decreasing BrdU labelling of A549 cells during the progression from subconfluence to day 5 postconfluence. Data are presented as the mean±sem of at least three cell cultures. Scale bars=50 μm **: p<0.01 versus SC by ANOVA and Newman–Keul's test. See text for details.](http://www.qdcxjkg.com/content/erj/42/1/198/F1.large.jpg?width=800&height=600&carousel=1){kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
![Downregulation of angiotensin-converting enzyme (ACE)-2 in proliferating epithelia of mouse lung instilled with keratinocyte growth factor (KGF) in situ. C57BL6 mice were administered purified recombinant KGF or vehicle (SHAM) intratracheally. 1 h before sacrifice, animals were administered bromodeoxyuridine (BrdU; 50 mg·kg−1 i.p.) for identification of proliferating cells [21]. Lung sections were immunolabelled with antibodies against ACE-2 (a, c and e) or BrdU (b and d). f) Lungs were homogenised for ACE-2 enzyme assay. Data are presented as the mean±sem of n=4 each for SHAM and KGF day 2. Scale bars=50 μm. FU: fluorescence units. *: p<0.05. See Materials and Methods for details.](http://www.qdcxjkg.com/content/erj/42/1/198/F4.large.jpg?width=800&height=600&carousel=1){kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}