





摘要
特发性肺纤维化(IPF)是成人最常见的慢性特发性间质性肺炎。法国的罕见病管理是由国家罕见病计划组织的,该计划支持在法国各地建立罕见病专家中心网络。本文概述了法国IPF管理指南的执行摘要,这是由法国国家参考中心和罕见肺病区域能力中心网络发起的一项倡议。本综述旨在为肺科医生提供以下文件:1)结合当前可用的证据;2)回顾IPF诊断和管理的实用模式;3)适应日常医疗实践。法国的实用指南是一个协调委员会、一个编写委员会和一个多学科审查小组共同努力的结果,并遵循了高级Autorité de Santé的建议。本文中包含的所有建议都得到了审查小组至少90%的同意。在此,我们总结了法国指南的主要结论和实际建议。
简介
特发性肺纤维化(IPF)是一种不明原因的纤维增生性不可逆疾病。IPF的发展通常是进行性的,主要发生在60岁以上,局限于肺部。它是成人慢性特发性间质性肺炎中最常见的类型。
IPF曾经被认为是一种孤儿病,因为没有被证明有效的特定治疗方法可用,IPF是一种罕见的疾病,在美国估计患病率为每10万人14至28例[1].美国每年的估计发病率为每10万人6.8至8.8例[1].法国尚未发表IPF的流行病学估计。
在法国,一个国家参考中心和9个罕见肺病区域能力中心得到批准,在两个国家罕见病计划(2008-2011年和2013-2016年)的总体框架内组织IPF的诊断和管理。
自2011年IPF诊断和管理国际指南出版以来[2],已经发表了新的数据,特别是关于几种新的治疗方法的疗效和耐受性,这些治疗方法旨在改变疾病的发展或缓解症状。目前的目标是,在参考资料和能力中心的协调下,为肺科医生提供当前可用数据的概要,并使用适应于现实生活日常实践的术语,尽可能清楚地定义IPF的诊断方式和以患者为中心的管理[3.].
本文件是执行摘要的英文版本,概述了诊断和管理指规性肾炎的法文指南的主要结论[4].
委员会的任务
本文由法国IPF专家撰写,作为2011年发表的IPF诊断和管理的国际建议的实用概述[2],并结合对2011年以来该领域发表的文献(包括治疗试验)的批判性回顾。这篇文章是由一个协调委员会,一个写作委员会和一个审查委员会。
委员会通过了适用于良好临床实践发展的规则,该规则是基于由高级卫生部Autorité de Santé [5].
协调委员会
协调委员会向Société de Pneumologie de languue Française (SPLF)提交了工艺和验证方案,对文献进行了系统审查,为编写委员会准备了文件的初稿,组织了工艺和验证方案并监测其应用情况,并向SPLF提交了经编写委员会和审查委员会验证的建议。
编写委员会
编写委员会对协调委员会编写的文件第一版进行了评估。他们采用三分制(我同意、我犹豫或我不同意)确定了需要审查的要点,对文件的形式和内容提出了建议,并对提交给审查委员会的文件进行了验证。
审查委员会
审查委员会由三名在大学医院工作的肺科医生(不包括能力中心)、三名在综合医院工作的肺科医生、三名在私人诊所工作的肺科医生、两名在间质性肺病方面具有专门知识的放射科医生和两名专攻胸科病理学的病理学家组成。委员会对所有主题和相应建议进行了评估,评分范围从1(完全不同意)到9(完全同意)。投票是通过电子方式进行的,结果在分析之前是匿名的。所有提交审核委员会审查的建议均已首先获得至少80%的撰写委员会成员批准[5].每个陈述的评级是基于文献中发表的数据的综合(与问卷一起提供)和读者在相应领域的经验。审查委员会成员只能回答他们认为有能力回答的问题。
指引发展过程
拟订这些建议包括以下步骤。1)对协调委员会自2010年以来在指规数领域发表的文献进行批判性审查。2)协调委员会初版。3)写作委员会对初版的评审。4)由协调委员会制作修订本。5)写作委员会评审并投票。6)对协调委员会未能达成共识的问题进行重写。7)评审委员会评审表决。8)由协调委员会制作修订本。9)由SPLF科学理事会审查。10)由协调委员会制作修订版。 11) Submission of the manuscript.
苏丹人民解放阵线科学理事会就这些建议的相关性、写作和适用性提出了意见。建议须获评审委员会至少90%的委员批准,方可生效[5].
这些建议拟定如下。1)“建议”是指所描述的选项与大多数患者相关(如。已确定疗效的治疗)。2)“建议”是指所描述的方案可能与某些患者相关(如。治疗很可能有效)。3)“有可能”是指所描述的选项可能与某些患者相关,但现有数据不允许给出更有力的建议(如。治疗效果不确定)。4)“不推荐”是指所描述的方案与大多数患者不相关(如。治疗被证明效率低下)。5)“It is recommended not to”是指应避免上述选项(如。有有害影响的治疗)。
SPLF科学理事会于2013年1月10日批准了用于制定这些建议的方法,并于2013年6月12日批准了书面建议。有关建议于2013年12月公布[4].在此,我们总结了法国指南的主要结论和实际建议。
IPF的诊断
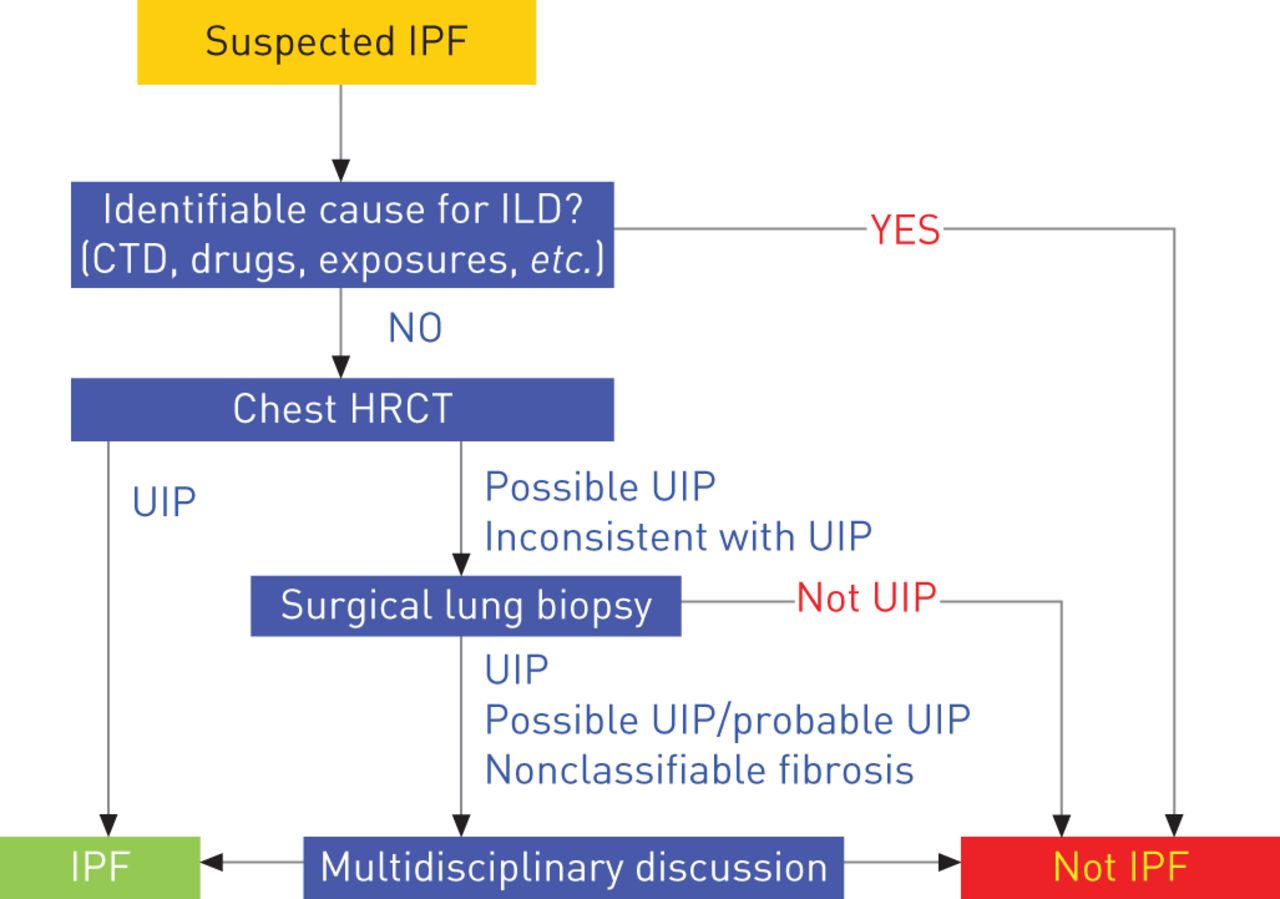
IPF是一种病因不明的纤维增生性疾病,与通常间质性肺炎(UIP)的组织病理学和/或高分辨率计算机断层扫描(HRCT)模式有关[2].存在放射学和/或组织学UIP模式是建立IPF诊断的必要条件。在未接受视频辅助手术肺活检(SLB)的患者中,如果HRCT显示(明确的)UIP模式(图1而且表1).在接受视频辅助SLB的患者中,在HRCT和SLB方面的特定组合显示UIP模式时,就可以确定诊断(表2).在所有病例中,需要排除ILD的其他已知原因(特别是与环境有关的原因,特别是职业暴露、药物毒性或全身性疾病),以确定IPF的诊断。

特发性肺纤维化的诊断算法。间质性肺疾病;CTD:结缔组织病;HRCT:高分辨率计算机断层扫描;UIP:普通间质性肺炎。转载自[2,并得到出版商的许可。
IPF主要发生于60至70岁之间,以男性略多见[6- - - - - -9].IPF没有特定的临床体征,这就解释了为什么诊断经常(太)晚。最初的临床表现为进行性劳力性呼吸困难并干咳;双基底吸气裂纹(魔术贴裂纹)是持续的,在疾病早期出现[2,10- - - - - -13].手指杵状畸形的发生率为~ 50%。体重减轻和一般状态的改变是不常见的。紫绀和右心室衰竭的体征仅发生在呼吸功能不全的晚期。病情发展为慢性限制性呼吸衰竭和死亡。血管前肺动脉高压常出现在晚期,特别是如果肺气肿合并IPF。
问题1:对于可能患有IPF的弥漫性间质性肺炎的患者,应该进行临床调查的主要原因是什么?
建议
建议对考虑诊断为IPF的患者进行临床调查,包括暴露于药物制剂、吸入有机抗原或矿物颗粒,或结缔组织疾病和癌症等ILD的原因。
评论
IPF的诊断需要排除其他形式的ILD [14],包括有明确病因的间质性肺炎:吸入有机抗原引起的过敏性肺炎、药物制剂的毒性、矿物质(二氧化硅和石棉等)引起的尘肺病、原发性或继发性癌症、创伤性或血流动力学性肺水肿;或2)病因不明,但发生在结缔组织疾病(特别是类风湿性关节炎、Sjögren综合征和系统性硬化症)、结节病、明确界定的浸润性肺部疾病,如淋巴管肌瘤病、肺朗格汉斯细胞肉芽肿病或特发性慢性嗜酸性粒细胞性肺炎,或任何其他明确识别的ILD。
问题2:IPF患者应该做什么生物检查?
建议
如果考虑IPF的诊断,建议对结缔组织病的生物学体征进行调查。
建议进行生物检查,包括以下确定。1)差异血细胞计数,c反应蛋白,血清肌酐,转氨酶,γ-谷氨酰转移酶和碱性磷酸酶。2)抗核抗体、抗瓜氨酸环肽抗体、类风湿因子。3)根据临床表现或是否检测到抗核抗体、Sjögren综合征特异性抗体(抗ssa、抗ssb)、系统性硬化症抗体(抗着丝粒、抗拓扑异构体酶-1、抗u3rnp)、抗合成酶抗体、抗甲状腺抗体、磷酸肌酸激酶、血清蛋白电泳。
评论
DIP与与UIP相容的放射学和/或病理模式相关[15],并可能是结缔组织病的首个临床表现[16].因此,如果考虑到IPF的诊断,有必要系统地观察肺外体征和生物标志物,以消除结缔组织疾病。如果在疾病过程中出现提示结缔组织疾病的体征、症状或生物学异常,应质疑IPF的诊断。还应测量炎症综合征或肺外功能障碍的生物学标记物。已提出检测抗中性粒细胞细胞质抗体[17].在暴露于有机抗原或怀疑过敏性肺炎的情况下,沉淀素检测是合理的。研究感染因子,特别是支气管肺泡灌洗(BAL),可能是合理的。如果怀疑是IPF以外的ILD,则应进行旨在确定淋巴增性疾病的检查(蛋白电泳、免疫电泳、尿免疫固定或冷球蛋白血症)。
问题3:怀疑患有IPF的患者是否应该进行BAL ?
建议
如果考虑IPF的诊断,建议进行BAL,特别是如果HRCT没有显示明确的UIP模式。
问题4:什么时候对怀疑患有IPF的患者进行基因检测?
建议
如果怀疑诊断为IPF,建议在医学访问期间系统地进行检查,以确定家族内ILD的其他原因,并寻找提示遗传原因的临床和生物学迹象(肝脏、皮肤、粘膜和血液系统异常)。
建议在家族背景下出现IPF的患者转到专门从事遗传学的门诊建立谱系,并建议主要针对目前可用的知识,端粒酶复合物基因和表面活性剂蛋白- c基因进行遗传分子分析。
问题5:诊断IPF时应做哪些肺功能检查?
建议
建议将肺活量(FVC)和肺对一氧化碳的扩散能力(DLCO)应在接受IPF测试的患者中进行评估。建议还应评估总肺活量、室内空气中静息动脉血气、6分钟步行试验(6MWT)距离和经皮氧饱和度。
评论
IPF患者静息时肺功能检查显示几种异常:1)肺功能受限模式(FVC和总肺活量降低);2)的早期减少DLCO肺对一氧化碳的转移系数;3)通常,静息时动脉血气值正常或低碳酸血症(肺泡-动脉氧张力差增大)。
此外,运动时进行的肺功能测试显示运动能力降低,可通过以下测量进行评估:运动低氧血症(即使休息时没有);6MWT行走的距离;运动时经皮氧饱和度降低,尤其是在6MWT期间;运动时最大摄氧量和最大功率负荷的降低。
问题6:在没有接受视频辅助SLB的患者中,HRCT何时足以诊断IPF ?
建议
建议HRCT表现为UIP模式,特别是蜂窝状模式(表2),如果排除了ILD的其他原因,则认为足以诊断IPF。
评论
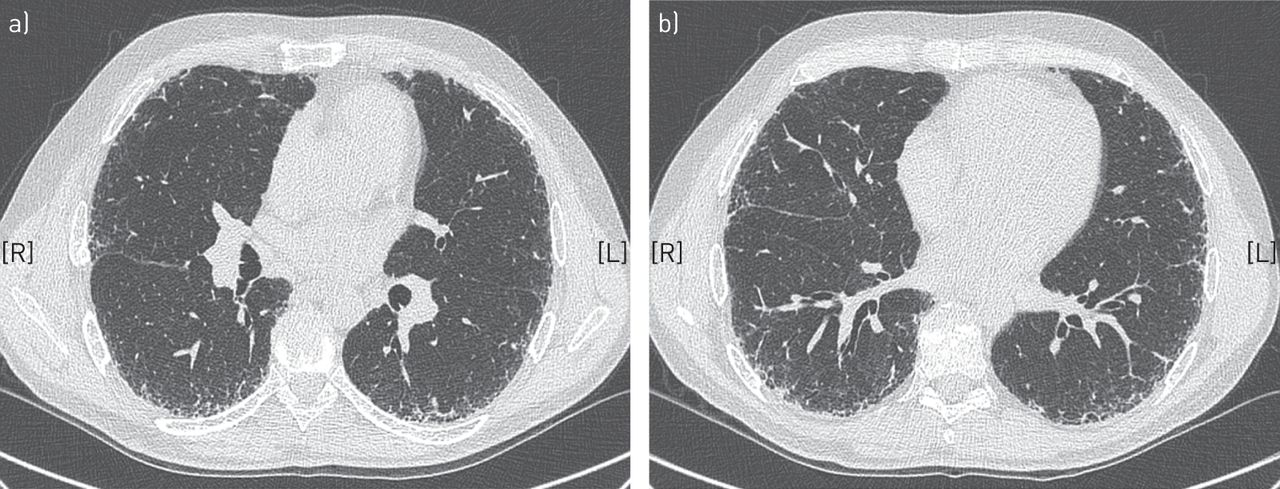
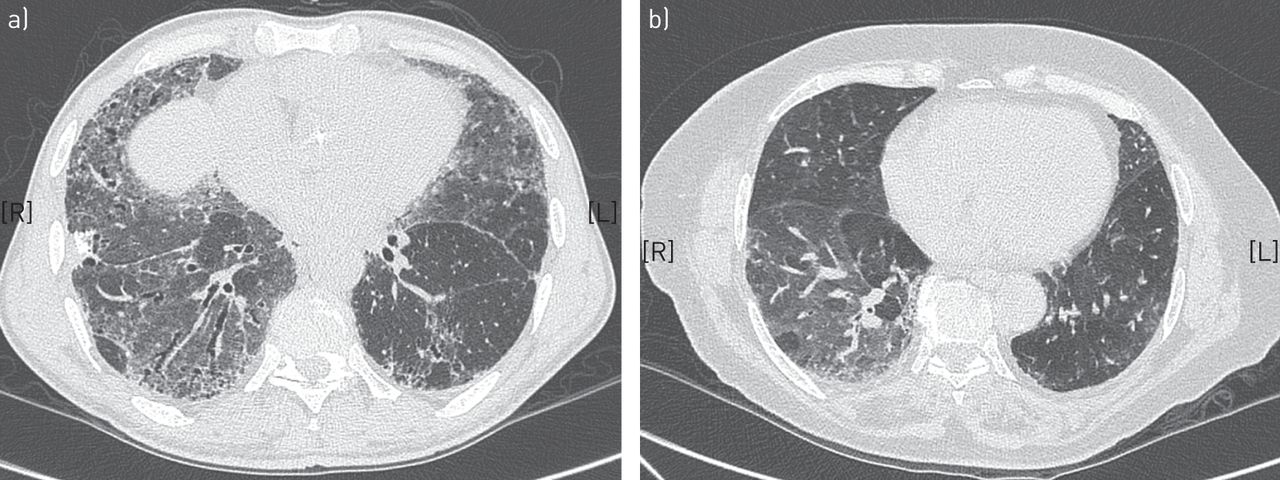
胸部HRCT是诊断IPF的必要手段。约50%的病例HRCT显示明确的UIP模式伴蜂窝状(图2) [2,28],如果分析是由在IPF领域有经验的肺科医生团队在兼容的临床环境下进行的,这就足以诊断IPF。在其他情况下,成像特征不具有特征性(图3)和视频辅助SLB来确定诊断。在某些情况下,HRCT表现可能与UIP模式不一致(图4).表2表示用于定义HRCT结果为确定或可能的UIP的标准。为了在HRCT上确定UIP模式,需要进行蜂窝蜂窝。表3指示如何进行HRCT。

高分辨率计算机断层扫描显示了典型的间质性肺炎,伴有蜂窝状改变和牵引性支气管扩张,以及胸膜下和基底网。未见提示其他诊断的特征。

a, b)肺活检的代表性例子。高分辨率计算机断层扫描显示可能常见的间质性肺炎模式,胸膜下和基底网,牵引性支气管扩张。未见提示其他诊断的特征。视频辅助肺活检显示明确的常见间质性肺炎类型。

高分辨率计算机断层扫描显示的模式与通常的间质性肺炎不一致。a)特发性非特异性间质性肺炎患者伴有牵引性支气管扩张的显著弥漫性磨玻璃衰减。b)超敏性肺炎患者毛玻璃衰减及马赛克分布。
问题7:哪些患者应该考虑SLB来确定IPF的诊断?
建议
对于怀疑IPF诊断的患者,如果HRCT上没有明确的UIP模式,建议考虑视频辅助SLB。在仔细评估手术风险后,多学科小组讨论决定进行活检。必须考虑与活检相关的潜在风险,特别是由于患者的年龄,合并症的存在,以及间质性肺炎的严重程度和进展。
问题8:多学科小组讨论在IPF诊断中的作用是什么?它是如何进行的?
建议
在涉及肺科医生、放射科医生和间质性肺炎领域经验丰富的病理学家的多学科讨论中,建议IPF的明确诊断应综合临床评估、计算机断层扫描特征以及(如果可能的话)病理特征(表1).
建议将复杂的病例转到专家中心(参考或主管中心),或转到在ild方面有经验的肺科。
评论
IPF的诊断综合了临床、放射学、生物学、呼吸功能和病理特征,并在多学科讨论中建立。理想情况下,多学科讨论在专家或专业中心进行,即。参考中心或其中一个罕见肺部疾病的能力中心,或有经验的肺科。
问题9:如何评估IPF患者的预后?
建议
建议IPF患者在诊断时根据以下因素评估预后:呼吸困难的严重程度;肺功能测试结果(FVC和DLCO);6MWT结束时经皮氧饱和度;HRCT上的蜂窝状程度;超声心动图显示肺动脉高压征象;使用分数。在随访期间,IPF患者的预后也应根据以下因素进行评估:症状进展;FVC和DLCO;HRCT显示纤维化;在适用的情况下,超声心动图显示肺动脉高压的迹象。
评论
以下因素与IPF患者死亡风险增加有关[29].1)年龄和男性。2)早期体征和症状:包括呼吸困难严重程度;DLCO预测值< 35-40%,室内空气下6MWT经皮氧饱和度<88%,HRCT显示蜂窝状程度,毛细血管前肺动脉高压。3)临床过程中出现的体征和症状:包括呼吸困难加重,6个月以上FVC下降>5%(绝对值)或10%(绝对值或相对值)[30.], >15%(绝对值或相对值)的下降DLCO超过6个月,在6MWT期间,>行走距离减少50 m [31], HRCT显示纤维化加重。
1年、2年和3年的生存估计可以使用基于年龄、性别、FVC和生理的GAP(性别、年龄、生理)评分来预测DLCO(www.acponline.org/journals/annals/extras/gap/) [32,33].
IPF的治疗
有哪些治疗IPF的药物建议?
强的松,硫唑嘌呤和N-乙酰半胱氨酸三联疗法
建议
建议不要开始强的松,硫唑嘌呤和三联治疗N-乙酰半胱氨酸(NAC)在确诊IPF患者中的作用。
评论
在IFIGENIA(特发性肺纤维化国际小组探索n -乙酰半胱氨酸I年度)随机、安慰剂对照试验中[34],抗氧化剂量(1.8克·天−1)可减少植被覆盖度的下降DLCO与安慰剂相比,同时接受强的松和硫唑嘌呤联合治疗的IPF患者。这项试验的目的不是评估三联疗法与安慰剂相比的益处。
PANTHER(强的松龙、硫唑嘌呤和NAC:一项评估IPF反应的研究)试验评估了NAC抗氧化治疗轻中度IPF的疗效[35].试验最初包括三个样本量相似的组(三联疗法:硫唑嘌呤+强的松+ NAC与南汽与安慰剂),主要结果是60周时FVC下降。与安慰剂相比,由于全因死亡风险增加(p=0.01)和非计划住院(p<0.001),三联治疗组在纳入236例患者6个月后过早停用。
南汽单药治疗
建议
考虑到收益/风险比和患者的偏好,如果在考虑参加治疗性临床试验后,不表明使用已批准的药物进行治疗,可以对一些明确诊断为IPF的患者开NAC治疗。
皮质类固醇治疗
建议
对于明确诊断为IPF的患者,建议不要使用皮质类固醇治疗(伴有或不伴有免疫调节剂治疗),除非在疾病急性加重的情况下。
抗凝治疗
建议
建议不要口服抗维生素K抗凝剂治疗IPF。虽然缺乏具体的数据,但如果IPF患者是出于其他原因(特别是心血管指征),口服抗维生素K药物并不禁忌。
秋水仙碱,环孢素A,干扰素-γ-1b和依那西普
建议
对于明确诊断为IPF的患者,不建议使用秋水仙碱、环孢素A、干扰素-γ-1b或依那西普治疗。
内皮素-1受体拮抗剂
建议
不建议明确诊断为IPF的患者使用波生坦或马西坦治疗。对于明确诊断为IPF的患者,建议不要使用氨布里森坦治疗。
评论
在波生坦的II期随机对照试验中,一种内皮素-1受体a和B的拮抗剂(ET一个和等B,分别),主要终点(改良的6MWT)无改善[54].BUILD(波生坦在间质性肺疾病中的应用)-3,在肺活检证实的IPF患者中进行[55],在波生坦组的主要终点(到IPF恶化或死亡的时间)或生活质量和呼吸困难方面没有显示出任何改善。评估马西坦的MUSIC(马西坦在特发性肺纤维化临床中的应用)试验也未达到主要终点(FVC变化)[56],另一种ET的拮抗剂一个和等B。在ARTEMIS-IPF中[57, ambrisentan, ET一个拮抗剂对主要终点(死亡时间或肺功能恶化)有不利影响,并与呼吸并发症住院率增加有关。安布里森坦目前是IPF的禁忌症,包括伴有严重肺动脉高压的IPF。
道,
建议
对于明确诊断为IPF的患者,不建议开依那西普治疗。
Pirfenidone
建议
目前建议治疗明确诊断为轻度至中度IPF的患者(定义为FVC≥50% pred和DLCO≥35% pred)与吡非尼酮。这种治疗应由在IPF诊断和管理方面有经验的医生发起和监督,并需要定期监测临床耐受性和肝酶。患者在吡非尼酮治疗期间不得吸烟,应警告患者避免紫外线照射。
评论
对轻度至中度IPF患者进行的两项III期研究(006/Capacity 1和004/Capacity 2)的汇总分析显示,使用吡非尼酮治疗的患者在72周时FVC下降显著降低[58].疾病进展(定义为FVC绝对值下降≥10%,FVC绝对值下降≥15%DLCO或死亡)和6MWT性能的下降也有所下降。在日本进行的III期研究中[59],吡非尼酮(1800毫克·天−1)与安慰剂相比显著降低了FVC的下降。另一项III期临床试验正在美国进行(www.clinicaltrials.gov;标识符NCT01366209).
吡非尼酮于2011年获得欧洲上市许可,并于2012年在法国上市,用于临床和放射学证实诊断为轻度至中度IPF (FVC≥50% pred和DLCOpred≥35%)。吡非尼酮应由在IPF诊断和管理方面有经验的专科医生启动和监督。
吡非尼酮不应用于氟伏沙明治疗的患者和严重肝或肾损害的患者。吡非尼酮最常见的不良反应为恶心、皮疹、疲劳、腹泻、消化不良、光敏反应和体重减轻[58].吡非尼酮也可诱导肝酶升高。在吡非尼酮治疗开始前应进行肝功能检查,随后前6个月每隔一个月进行一次,此后每3个月进行一次。
吸烟有可能增加参与吡非尼酮代谢的酶的活性,在吡非尼酮治疗前和治疗期间应停止吸烟。同时使用奥美拉唑理论上可能导致吡非尼酮药代动力学的变化,应该避免。
IPF患者是否应接种流感和肺炎球菌感染疫苗?
建议
建议明确诊断为IPF的患者每年接种流感疫苗和抗肺炎球菌疫苗。
评论
虽然在IPF的背景下没有对这些疫苗进行具体的研究,但很有可能,就像其他患有慢性呼吸道疾病的患者一样,IPF患者如果出现肺炎球菌感染或流感,就会暴露在高风险中。抗肺炎球菌疫苗可使用多糖肺炎球菌疫苗进行。
IPF患者应该接受补充氧疗吗?
建议
建议明确诊断为IPF和休息时严重低氧血症(严重慢性呼吸衰竭)的患者使用长期氧治疗。
IPF患者应该接受呼吸康复吗?
建议
建议在明确诊断为IPF的运动能力限制导致严重损害的患者中启动呼吸康复计划。
IPF患者肺移植的适应证是什么?
建议
如果疾病严重或恶化,建议所有明确诊断为IPF的<65岁患者考虑肺移植。建议患者在病程早期获得肺移植的相关信息。建议在肺移植中心对患者进行早期评估。
IPF急性加重的诊断标准是什么?
建议
在排除了其他可能导致呼吸功能恶化的原因(即。感染、肺栓塞和左心衰竭)。
评论
IPF急性加重的特征是急性(<30天)呼吸困难加重,但无明确原因(如。感染,肺栓塞,左心衰或心律失常)的患者明确诊断为IPF。HRCT除原有异常外,还显示新的不透明,尤其是磨玻璃样不透明。低氧血症恶化是常见的(≥10 mmHg降低PaO2).视频辅助SLB通常被认为太危险,在这种情况下不执行。
IPF急性加重患者可建议哪些治疗方法?
建议
建议使用高剂量皮质类固醇治疗IPF急性加重。静脉注射环磷酰胺治疗IPF急性加重是可能的。关于使用低分子肝素治疗IPF急性加重的数据不足。
有创通气和无创通气在IPF管理中的作用是什么?
建议
对于明确诊断为IPF和急性或慢性呼吸衰竭的患者,不建议使用有创通气。在少数IPF和急性呼吸衰竭患者中使用有创或无创通气是可能的;特别是,如果符合紧急肺移植的标准,如果急性加重是IPF的首要表现,或者在急性感染或可逆原因的情况下。
是否应该对IPF患者进行肺动脉高压调查,如果应该如何调查?
建议
建议在IPF诊断过程中进行超声心动图检查,然后每年检查IPF患者的肺动脉高压。对于明确诊断为IPF的患者,建议行右心导管术诊断肺动脉高压:1)肺移植前;2)临床恶化,运动能力受限,运动能力下降DLCO(特别是DLCO<40% pred)和/或低氧血症与限制性通气缺陷(特别是如果存在肺气肿)不成比例;3)认为有必要对预后进行准确评估;4)超声心动图怀疑重度毛细血管前肺动脉高压(三尖瓣反流>3.5 m·s−1),讨论肺动脉高压的适应症外治疗方法;5)怀疑左心室疾病但收缩功能保留。
如果IPF患者存在肺动脉高压,应如何处理?
建议
建议对患有IPF和肺动脉高压的患者进行静息性低氧血症、血栓栓塞性静脉疾病和左心衰竭的调查,并在需要时对患者进行适当的治疗。也应该考虑肺移植。
在IPF和中度毛细血管前肺动脉高压患者(平均P巴勒斯坦权力机构≤35 - 40mmhg),不建议对肺动脉高压进行任何特定的治疗。
在IPF和严重的毛细血管前肺动脉高压患者(平均P巴勒斯坦权力机构≥35-40 mmHg),如果肺动脉高压被认为是导致症状恶化的原因,则有可能对肺动脉高压开出特定的治疗方案,最好是在专门的中心使用西地那非。
在IPF和严重的毛细血管前肺动脉高压患者(平均P巴勒斯坦权力机构≥35-40 mmHg),建议不要开氨布里森坦。
评论
除IPF外,肺动脉高压的原因应始终加以探讨。根据年龄和共病情况,应考虑肺移植或心肺移植。对于IPF患者,没有推荐或批准针对肺动脉高压的特定治疗方法[83].
在对一些IPF和肺动脉高压患者进行的两项非对照前瞻性西地那非研究中,在治疗8-12周后,步行距离和肺血流动力学得到改善[88,89].在随机对照的STEP-IPF(西地那非在IPF中的运动表现试验)试验中,将西地那非治疗和安慰剂进行了12周的比较,没有观察到6MWT(主要终点)的显著改善[90],但西地那非显著改善了动脉氧合,DLCO,呼吸困难和生活质量,并可用于超声心动图右心室功能障碍患者[91].
一项随机对照试验,ARTEMIS-IPF (www.clinicaltrials.gov;标识符NCT00768300) [57,显示出一种ET——氨布里森坦的有害作用一个拮抗剂,主要终点(死亡时间或呼吸功能恶化)和因呼吸并发症住院人数增加。这导致阿姆布里森坦在IPF中的另一项试验,ARTEMIS-PH (www.clinicaltrials.gov;标识符NCT00879229).因此,安布里森坦是IPF患者的禁忌症。riociguat在ILD患者中的开放标签研究提示心排血量增加,但临床获益不明确[92].
是否应该对IPF患者进行胃食管反流调查,如果应该如何调查?IPF患者胃食管反流应如何处理?
建议
建议在所有明确诊断为IPF的患者的医学访谈中调查胃食管反流的病史或症状。如果怀疑胃食管反流,建议进行适当的检查,并遵守胃食管反流管理的适用建议。
IPF患者是否需要检查肺气肿,如果需要,如何检查?IPF患者肺气肿应如何处理?
建议
建议在IPF诊断时在胸部HRCT上寻找肺气肿,以免在保留肺容量的情况下低估疾病的严重程度。建议如果存在肺气肿,应类似于其他情况下的肺气肿,包括戒烟,如果存在气流阻塞,应使用吸入性支气管扩张剂。目前尚无数据支持联合肺纤维化和肺气肿的特定药物治疗。
评论
与无肺气肿的IPF患者相比,合并肺纤维化和肺气肿患者的肺呼吸量保留,一氧化碳转移减少,需氧量增加。与单纯IPF患者相比,该综合征患者的疾病进化可能更糟[One hundred.,101],但没有可靠的数据表明肺气肿会影响生存率。联合肺纤维化和肺气肿的诊断很重要,这样就不会将肺容量的保留错误地归因于疾病的轻微程度。肺动脉高压尤其常见,是该综合征患者死亡的主要预测因素[102].可能需要长期氧疗。监测植被覆盖度和DLCO不能准确评估预后[103,104].
目前尚无数据支持对IPF患者进行特定的肺气肿治疗,或对合并肺纤维化和肺气肿患者进行特定的纤维化治疗的建议。可能的治疗方法(单药治疗中的吡非尼酮和NAC)必须单独评估,考虑到副作用,在该适应症中缺乏这种治疗的潜在益处的数据,以及评估疾病演变的困难(FVC变化很小)。
是否应该对IPF患者进行阻塞性睡眠呼吸暂停综合征的检查,如果需要,如何检查?
建议
如果临床体征提示阻塞性睡眠呼吸暂停综合征,建议明确诊断为IPF的患者进行通气测谎。
IPF患者应如何处理阻塞性睡眠呼吸暂停综合征?
建议
目前尚无IPF患者睡眠呼吸暂停治疗的具体数据。有人建议,当确诊为IPF的患者出现阻塞性睡眠呼吸暂停综合征时,应按照适用于无IPF患者的通常建议进行管理。
评论
现有数据不足以推荐IPF患者的阻塞性睡眠呼吸暂停综合征的特定治疗方法[109].
IPF患者的症状应如何处理?
建议
有可能在出现可待因不能缓解的干咳的IPF患者中开一种短暂的、低剂量的口服皮质类固醇治疗。应监测该疗法的疗效和耐受性。
对于明确诊断为IPF并表现为运动时严重呼吸困难和运动时氧饱和度降低(日常生活活动或标准化运动时经皮氧饱和度<88%,如6MWT)的患者,可以开门诊补充氧治疗。
在没有高碳酸血症的情况下,有可能在出现严重呼吸困难的IPF患者中使用低剂量吗啡衍生物,同时监测这种治疗的疗效和耐受性。
评论
有限的数据表明,口服皮质类固醇治疗和沙利度胺可以缓解IPF相关的慢性咳嗽。高剂量皮质类固醇或沙利度胺耐受性差,不可取。低剂量吗啡衍生物可用于严重呼吸困难的病例,但应密切监测这种治疗的副作用[110].低剂量吗啡衍生物(每天≤30mg口服吗啡当量)与慢性阻塞性肺病患者开始长期氧疗的死亡率增加无关[111].在IPF患者中没有类似的数据。
一项小型研究表明,接受氧气治疗的IPF和休息时低氧血症患者的运动能力可能得到改善[112].两项回顾性研究表明,动态氧疗可显著改善IPF患者的6MWT表现和呼吸困难[113,114].在这两项研究中,氧流量逐步增加,直到一项研究经皮氧饱和度≥88%,另一项研究≥90%。
在IPF患者的随访过程中,应该进行哪些检查以及以何种频率进行检查?
建议
建议明确诊断为IPF的患者应进行临床访问并进行肺功能检查,包括每3-6个月测量一次FVC。建议进行胸部计算机断层扫描:如果怀疑IPF急性加重;发生不明原因的临床改变;如怀疑患有肺癌;在肺移植之前。
IPF患者随访期间肺癌风险是否增加?
建议
IPF患者患肺癌的风险增加。建议让负责随访的医生了解明确诊断为IPF的患者经常发生肺癌。建议敦促吸烟者戒烟,并告知戒烟援助。对于明确诊断为IPF和肺癌的患者,建议在治疗决策时考虑IPF。
IPF患者是否应调查其他共病?
建议
建议在明确诊断为IPF的患者中评估共病情况(心血管疾病、血栓栓塞性静脉疾病、糖尿病和抑郁症),以便通知负责随访的医生。
评论
IPF患者常伴有共病,特别是与吸烟有关的疾病,包括心血管疾病、血栓栓塞性静脉疾病、糖尿病和抑郁症[120].现有的数据不足以建议对这种共病进行系统筛查,但告知负责随访的医生(负责护理组织的全科医生或肺科医生)共病的存在是重要的。
结论
自从关于IPF诊断和管理的ATS/ERS/JRS/ALAT指南于2011年发布以来,来自主要临床研究的新数据促使几个欧洲国家的IPF专家以国家共识声明或意见的形式发布了更新的建议[121,122].
需要将最近的证据纳入诊断和管理IPF的建议是本文件的基础。我们的主要目标是为法国肺科医生提供一种最新的、实用的决策工具,可用于怀疑或明确诊断为IPF的患者的日常临床护理。
本文件符合特定的国家方法和验证过程,由高级Autorité de Santé定义为指南制定的良好临床实践。这三个具有特定任务的委员会的组织确保了在诊断和/或管理IPF方面经验丰富的著名肺科医生、放射科医生和病理学家的参与。
在法国,参考中心和罕见肺病能力中心网络负责与当地肺科医生、普通医生和参与诊断和管理的其他专家合作,组织罕见病患者的护理,包括IPF。这些专家中心在地理上分布在法国所有地区。参考中心和罕见肺病主管中心协调了本指南的整个编制过程,以便使护理标准化和优化,但本建议准确地反映了所有有关专家的集体想法。
本文件的另一个特点是,即使缺乏令人信服的支持(或反对)检查或治疗的证据,它也能给临床医生提供指导。诸如“建议……”或“有可能……”等表述,并附有证明这些表述的文献摘要,应有助于读者做出知情的临床判断,并决定在给定的临床情况下,相关的诊断或治疗干预是否合适。
我们的建议与2011年指南之间的主要差异来自于最近出现的证据。1) IPF患者不应使用强的松、硫唑嘌呤和NAC的三联治疗,因为它已被证明会增加死亡率。2)皮质类固醇治疗与大量发病率相关,与免疫调节剂治疗联合可能会增加死亡率。皮质类固醇治疗不再是治疗IPF的一种选择,除非是缓解无力性咳嗽或治疗IPF急性加重。3)安布里森坦目前是治疗IPF的禁忌症。4)迄今为止,吡非尼酮是唯一一种对IPF患者证明有效的药物治疗,也是唯一一种已获批准并获得上市许可的治疗方法。
我们有信心,目前的建议将帮助护理IPF患者的临床医生在急需的最新国际指南发布之前,将他们的实践基于现有的证据。
致谢
作者的隶属关系如下。V. Cottin:里昂平民收容所,Hôpital Louis Pradel,肺气科服务-中心référence国家罕见肺炎,里昂,Université里昂,Université克劳德·伯纳德·里昂1,INRA, UMR754 IFR 128,里昂,法国。B. Crestani:法国巴黎,CHU Bichat,罕见肺病能力中心,肺气科。D. Valeyre:肺气系,Hôpital阿维森纳大学,博比尼医学院,罕见肺病中心,博比尼,法国。B. Wallaert:里尔CHU,罕见肺病能力中心,里尔,法国。J. Cadranel: CHU Tenon,罕见肺病能力中心,法国巴黎。j。Dalphin: CHU Besançon,罕见肺部疾病能力中心,Besançon,法国。P. Delaval:法国雷恩罕见肺病能力中心的临床医生。D. Israel-Biet: CHU Georges Pompidou,罕见肺病能力中心,法国巴黎。 R. Kessler: CHU of Strasbourg, Competence Centre for Rare Lung Diseases, Strasbourg, France. M. Reynaud-Gaubert: CHU of Marseille, Competence Centre for Rare Lung Diseases, Marseille, France. B. Aguilaniu: Clinique du Mail, Grenoble, France. B. Bouquillon: Opened Mind Health, Roubaix, France. P. Carré: Polyclinique Montréal, Carcassone, France. C. Danel: CHU Bichat, Paris, France. J-B. Faivre: Hôpital Albert Calmette, Lille, France. G. Ferretti: Hôpital Albert Michallon, Grenoble, France. N. Just: Centre Hospitalier Victor Provo, Roubaix, France. S. Kouzan: Centre Hospitalier Intercommunal sud Léman, Saint-Julien-en-Genevois, France. F. Lebargy: Hôpital Maison Blanche, Reims, France. S. Marchand-Adam: CHU de Tours, Service de Pneumologie, CEPR, Inserm U1100, Tours, France. B. Philippe: Centre Hospitalier René Dubos, Pontoise, France. G. Prévot: Hôpital Larrey, Toulouse, France. B. Stach: Cabinet medical, Valenciennes, France. F. Thivolet-Béjui: Centre de biologie et pathogie est, Hôpital Louis Pradel, Lyon, France. J-F. Cordier: Hospices Civils de Lyon, Hôpital Louis Pradel, Service de pneumologie – Centre de référence national des maladies pulmonaires rares, Lyon, France.
委员会成员如下:协调委员会:V. Cottin, B. Crestani, D. Valeyre, B. Wallaert, J-F Cordier。写作委员会:V. Cottin, B. Crestani, D. Valeyre, B. Wallaert, J-F。科迪尔,j·卡德拉内尔,j·c。Dalphin, P. Delaval, D. Israel-Biet, R. Kessler和M. Reynaud-Gaubert。评审委员会:由编审委员会成员增设以下人员。私人执业肺科医生:P. Carré, B. Stach和B. Aguilaniu。普通医院肺科医生:贾斯特、库赞和菲利普。大学医院肺科医生:F. Lebargy, S. Marchand-Adam和G. Prévot。放射科医生:G. Ferretti和J-B。 Faivre. Pathologists: C. Danel and F. Thivolet-Béjui.
我们得到了open Mind Health (Roubaix,法国)和Michel Bordier (Clamart,法国)的支持。
脚注
支持声明:我们得到了里昂1号慈善基金会(法国维勒班)的资金支持。
利益冲突:可以在本文的在线版本中找到信息披露err.ersjournals.com
来源:已提交的文章,同行评议。
- 收到了2014年3月4日。
- 接受2014年3月17日。
- ©2014人队
ERR文章是开放获取的,并根据188滚球软件创作共用属性非商业许可4.0。
参考文献










![Diagnostic algorithm for idiopathic pulmonary fibrosis (IPF). ILD: interstitial lung disease; CTD: connective tissue disease; HRCT: high-resolution computed tomography; UIP: usual interstitial pneumonia. Reproduced from [2] with permission from the publisher.](https://err.ersjournals.com/content/errev/23/132/193/F1.large.jpg?width=800&height=600&carousel=1){kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
跳转到
- 文章
- 摘要
- 简介
- 委员会的任务
- 指引发展过程
- IPF的诊断
- 问题1:对于可能患有IPF的弥漫性间质性肺炎的患者,应该进行临床调查的主要原因是什么?
- 问题2:IPF患者应该做什么生物检查?
- 问题3:怀疑患有IPF的患者是否应该进行BAL ?
- 问题4:什么时候对怀疑患有IPF的患者进行基因检测?
- 问题5:诊断IPF时应做哪些肺功能检查?
- 问题6:在没有接受视频辅助SLB的患者中,HRCT何时足以诊断IPF ?
- 问题7:哪些患者应该考虑SLB来确定IPF的诊断?
- 问题8:多学科小组讨论在IPF诊断中的作用是什么?它是如何进行的?
- 问题9:如何评估IPF患者的预后?
- IPF的治疗
- 结论
- 致谢
- 脚注
- 参考文献
- 图表和数据
- 信息和指标