





摘要
我们假设在从慢性阻塞性肺疾病(COPD)的吸烟者的气道平滑肌(ASM)细胞于香烟烟雾的响应将是来自吸烟者本质不同而不COPD,产生与气道重塑更大促炎介质和因素。
从吸烟者具有或不COPD获得ASM细胞,然后用香烟烟雾提取物(CSE)或转化生长因子β1刺激。生产趋化因子和基质金属蛋白酶(MMPs)的ELISA法测定,和胶原蛋白的细胞外基质ELISA的沉积。通过甲苯胺蓝附着和细胞示踪绿伤口愈合测定法测得的CSE的上细胞附着和伤口愈合的影响。
CSE增加CXCL8和CXCL1的来自人ASM细胞释放,并从与COPD的吸烟者细胞产生更多的CSE诱导CXCL1。生产MMP-1,-3和-10的,和胶原蛋白VIIIα1(COL8A1)的沉积分别增加了CSE,尤其是在其具有更高的生产MMP-1的和COL8A1的沉积COPD组。CSE降低ASM细胞附着和仅COPD组中伤口愈合。
从COPD吸烟者ASM细胞对CSE刺激,这可以部分解释,为什么有些吸烟者制定COPD更敏感。
摘要
香烟烟雾提取物诱导气道重塑相关的COPD患者气道平滑肌细胞的变化http://ow.ly/wK0ey
介绍
慢性阻塞性肺疾病(COPD)是全世界发病率和死亡率的主要原因,其造成的经济和社会负担不仅巨大,而且还在不断增加[1],[2]。在慢性阻塞性肺病中,肺部会发生许多变化,即出现持续性炎症和不可逆转的气流受限[3]。
气流受限是由三个相互关联的过程引起的:小气道壁的增厚(重塑)、小气道的丧失和肺气肿。然而,小型气道重构被认为对气流限制的影响最大[4] - [6]。此外,小气道重构可能是COPD的主要病理损伤。在一项使用慢性阻塞性肺疾病(COPD)肺组织的微计算机断层扫描分析的研究中,发现末梢细支气管的重塑和消失在显微镜下先于肺气肿的改变[7]。小气道在COPD重塑由折叠粘膜,基底膜和结缔组织的沉积增厚,以及气道平滑肌(ASM)作为质量增加,特别是在严重的慢性阻塞性肺病[6],[8],[9]。结缔组织包括细胞外基质(ECM)蛋白的缠结框架,和特定ECM蛋白已知在气道被改变的COPD患者[10],[11]。
在发达国家,慢性阻塞性肺病的发展的主要危险因素是吸烟。通过使用两个在活的有机体内和在体外模型吸烟对慢性阻塞性肺病病因学的影响已开始被了解。大多数研究表明COPD的病因学是吸烟引起的炎症导致组织损伤;然而,以往的研究表明,气道重构可能与炎症无关[12]。先前已发现,来自患有COPD的成纤维细胞响应于香烟烟雾提取物(CSE)产生纤连蛋白和基底膜蛋白聚糖或蛋白聚糖减少(核心蛋白聚糖和糖链蛋白聚糖)的过量,相对于从人细胞而不COPD [12],[13]。慢性阻塞性肺病的上皮细胞也不同,以CSE [回应14],但COPD ASM细胞是否CSE有不同的反应不得而知。
在这项研究中,我们假设慢性阻塞性肺病患者的ASM细胞对香烟烟雾的反应与非慢性阻塞性肺病患者的ASM细胞的反应具有本质上的不同,特别是在促炎介质的产生和与气道重塑相关的因素方面。
材料与方法
研究对象
获得主题信息有关诊断,吸烟史,肺功能。不包括与哮喘,感染性疾病或间质性肺疾病的诊断的受试者。根据气流受限的严重程度如下从谁被归类受试者获得样品[15]。1)非COPD:N = 21,用力呼气容积1秒(FEV1)/用力肺活量(FVC)≥70%和FEV1≥80%。2)COPD:N = 20,FEV1/ FVC <70%。在补充材料(表S1)提供全部细节。所有研究对象或近亲签署知情同意书。使用人类的肺组织中的所有实验的批准是由西南悉尼地区卫生服务,圣文森特医院悉尼,悉尼人类研究伦理委员会的大学伦理审查委员会(所有悉尼,澳大利亚)提供。
细胞培养和样品制备
人的ASM细胞是从人的肺中获得的,其方法经过了先前所述方法的改进[16]。人类ASM细胞从大约六阶或更大支气管显微切割,和最初在包含DMEM(Invitrogen公司,卡尔斯巴德,加利福尼亚州,美国),补充了5%胎牛血清(DKSH,墨尔本,澳大利亚),1%的生长培养基中培养抗生素(Invitrogen公司)和25mM HEPES(Invitrogen)中。所有的细胞支原体的存在检测为阴性分别设置才开实验,和通道2和7 ASM小区之间,使用在六孔或96孔培养板(BD Biosciences公司,北莱德,澳大利亚)接种在1×10的密度4细胞·厘米-2在生长培养基中在37℃/ 5%CO并孵育2细胞在含有0.1%胎牛血清、1%抗生素和25mm Hepes的DMEM培养基中饥饿72h,然后给予CSE或10ng·mL的刺激-1转化生长因子(TGF) -β1(研发系统,明尼阿波利斯,美国)在静默中。刺激后,收集人ASM细胞(6孔板)上清液,用0.016 mM氢氧化铵(NH)裂解细胞(96孔板)4OH)在37℃下20分钟,然后用0.05%PBS-吐温(TW)20(体积/体积)洗涤。这些上清液和无细胞的ECM板储存在-20℃直至分析。
香烟烟雾提取物
我们使用万宝路红香烟(菲利普莫里斯,维多利亚,澳大利亚),并且每个香烟含有1.1毫克的尼古丁,15毫克焦油,和15mg一氧化碳。CSE的制备是通过从一个先前描述的修饰的方法[12]。简单地说,一个万宝路香烟通过25mL的DMEM中以恒定的速率鼓泡,并将该溶液被认为是100%的浓度CSE。100%CSE中新鲜每次实验产生的,并稀释至最终工作浓度和30分钟内使用。
细胞活力和毒性测定
人类ASM细胞接种在96孔板中接种如上所述,并且细胞用CSE的系列稀释液从0.05%至50%。活细胞的线粒体活性通过Thaizolyl蓝四唑溴化物(MTT)(西格玛奥德里奇公司,圣路易斯,MO,USA)测定法进行测试。细胞的膜完整性通过乳酸脱氢酶(LDH)(Sigma Aldrich公司)测定,根据释放到培养基中的LDH细胞质的量进行测试。刺激72小时后,MTT和LDH释放使用分光光度计(SPECTRAMAX M2; Molecular Devices公司,桑尼维尔,CA,USA)测量与设定的吸光度分别为570nm / 690 nm和490nm处/ 690纳米。
趋化因子ELISA
人类ASM细胞在6孔板中接种如上所述,并且细胞用不同浓度的CSE或10ng·毫升-1TGF-β1的h。72的浓度CXCL8(白介素(IL) 8),处于受控(GROα)CCL2, CCL5和CXCL10上层清液从人类ASM细胞测量通过使用商业载人CXCL8 /引发,处于受控/ GROαCCL2 /单核细胞趋化蛋白1,CCL5 /咆哮,CXCL10 /诱导protein-10 ELISA试剂盒(研发系统)根据制造商的指示。使用分光光度计(Spectramax M2)在450 nm/570 nm处读取吸光度。
转录因子核因子κB和激活蛋白-1活性测定
如前所述,将人ASM细胞接种于6孔板中,用CSE(5%和10%)或10ng·mL刺激细胞-1TGF-β160分钟,然后核提取物被收集。核因子的活性(NF)-κB和激活蛋白(AP)-1各样品根据生产商的说明书,使用TRANSAM ELISA试剂盒(Active Motif的,卡尔斯巴德,CA,USA)进行了评估。吸光度使用分光光度计(SPECTRAMAX M2)在450nm / 655nm的读取。
实时PCR数组
从与吸烟者获得的人ASM细胞(n = 3)和不COPD(N = 3)刺激在体外用5%CSE,10%CSE或10ng·毫升-1的TGF-β1。RNA收集在48小时,然后使用分离RNA迷你试剂盒(生物在线,伦敦,英国)纯化,和mRNA,使用M-MLV逆转录酶(Invitrogen)转化成cDNA。等量来自相同组汇集各样品的cDNA的;然后将基因表达使用根据制造商的说明(Invitrogen)中的TaqMan阵列人胞外基质分子和粘附分子的96孔板进行测试。实时PCR使用的StepOne加检测系统进行,收集并通过的StepOne软件(Applied Biosystems,墨尔本,澳大利亚)的分析数据。mRNA的相对丰度是使用ΔΔCT方法[计算17]和结果进行标准化,以18S rRNA基因。
基质金属蛋白酶ELISA
人类ASM细胞在6孔板中接种如上所述,并且细胞用不同浓度的CSE或10ng·毫升-1TGF-β1 72 h。总浓度的基质金属蛋白酶(MMP) 1, 2, 3, -10年和-12年的测量使用人类MMP的ELISA试剂盒(研发系统)根据制造商的指示,执行和阅读使用Luminex分析器(Luminex 200系统;Luminex,布里斯班,澳大利亚)。MMP-1酶活性测定采用人源MMP-1荧光检测试剂盒(R&D系统),相对荧光单位采用激发波长320 nm、发射波长405 nm的荧光平板阅读器(Spectramax M2)测定。
ECM ELISA
人类ASM细胞接种在96孔板中接种如上所述,并且细胞用不同浓度的CSE的或10ng·毫升-172 h的TGF-β1。ECM游离盘子是用来衡量蛋白质的沉积在ECM ELISA根据之前修改的方法16]。用于检测ECM蛋白第一抗体是兔多克隆抗人抗体COL5(2微克·毫升-1)(Abcam公司,剑桥,英国),兔多克隆抗人胶原蛋白VIIIα1(COL8A1)抗体(2微克·毫升-1)(罗福斯生物制剂,利特尔顿有限公司,美国),小鼠单克隆反纤连蛋白抗体(2μg·毫升-1)(Millipore公司,Billerica的,MA,USA),和小鼠单克隆抗 - 人抗体串珠素(2微克·毫升-1)(表达载体)。兔免疫球蛋白(Dako Cytomation,斯特鲁普、钙、美国)同形像控制抗体和鼠标IgG1κ同形像控制抗体(BD生物科学)在同一浓度作为主要抗体。COL8A1 perlecan,测量的生物素化的山羊anti-rabbit抗体0.5μg·毫升-1和生物素化的鸡抗小鼠抗体0.8微克·毫升-1分别使用。
蛋白质印迹
人类ASM细胞被播种在six-well板块如前所述,和细胞被刺激CSE(5%或10%)或72年TGF-β1 h。每个样本收集评估的上层清液可溶性COL1和溶纤连蛋白使用西方的污点。蛋白质大小分级10%聚丙烯酰胺凝胶,转移到5%的聚偏二氟乙烯膜和阻塞(wt /卷)脱脂牛奶1 h。膜解决方案与主要孵化抗体(8.8μg·毫升-1毫升小鼠单克隆抗COL1抗体(Sigma Aldrich公司)或1微克·-1鼠单克隆反纤连蛋白抗体(微孔)在2%的牛血清白蛋白/ TBS-Tw) 2 h,其次是孵化与二次抗体(2.6μg·毫升-1兔抗小鼠Ig辣根过氧化物酶抗体(Dako公司Cytomation公司)在2%牛血清白蛋白/ TBS-TW)1个小时。使用的Immobilon西方Chemluminescent HRP Subtrate(Millipore公司)进行免疫印迹检测,并使用柯达图像站4000 MM(Eastman Kodak公司,纽约,NY,USA)进行分析带。每个样品中存在的蛋白质的量被确定为光密度的密度。
免疫组织化学
免疫组织化学样品和免疫组织化学方法的制备先前已经描述[18]。气道切片处理以最小化非特异性背景染色,并与初级兔多克隆抗人抗体COL8A1 1微克·毫升培养-1(Abcam公司)和兔IgG同种型对照抗体1微克·毫升-1(DAKO Cytomation公司)。的共轭次级抗体,标记的聚合物的抗兔(的EnVision +系统-HRP; Dako公司Cytomation公司)和组织染色用色原底物,液体DAB(Dako公司Cytomation公司)可视化。每个区段(每个受试者一个部分)的10个图像拍摄,并使用软件斐济(ImageJ的)免疫染色进行定量19]。补充材料中提供了详细信息。
细胞附着测定
96孔培养板暴露于生长培养基中72小时,并于24小时,然后暴露于72小时的不同浓度的CSE(0.05%至10%)的静止介质的静止平台。人类ASM细胞以5×10孔的密度接种在这些处理的板4细胞·厘米-2在2小时的静止平台。如先前所描述的细胞附着用甲苯胺蓝附着法检测[20.]。使用分光光度法在595nm处(SPECTRAMAX M2)的吸光度,测定附着的细胞的相对数目。补充材料中提供了详细信息。
伤口愈合分析
我们使用一个Oris细胞迁移组装包(Platypus Technologies, Madison, WI, USA)来进行伤口愈合试验。伤口是在处理过的96孔黑色板上造成的,细胞用细胞追踪器绿色CMFDA (Invitrogen)标记。将标记的人ASM细胞以5×10的密度接种于损伤的黑板上4细胞·厘米-2在生长介质。粘附24小时后,将塞子取出,孵育4小时。使用荧光平板阅读器(Wallac VICTOR)测量创面愈合值2;Perkin Elmer, Waltham, MA, USA)从底部读取,激励波长设置为485 nm,发射波长设置为535 nm。补充材料中提供了详细信息。
统计分析
数据分析使用GraphPad Prism 5.0软件(GraphPad software, San Diego, CA, USA)。所有数据均以均数±表示SEM。单因素方差分析,双向ANOVA加Bonferroni事后检验或Mann-Whitney检验被用作适当确定统计学意义。p值≤0.05被认为是显著。
结果
CSE对ASM细胞毒性
由于高浓度的CSE的已知是细胞毒性,我们使用两种毒理学试验,线粒体活性的评估通过MTT法和膜的完整性通过释放LDH,以确保我们使用的CSE浓度对人ASM细胞没有毒性作用。浓度≥15%时,CSE显著降低生存能力(图1);因此,我们在后续的实验中使用0.05%到10%的CSE作为刺激。
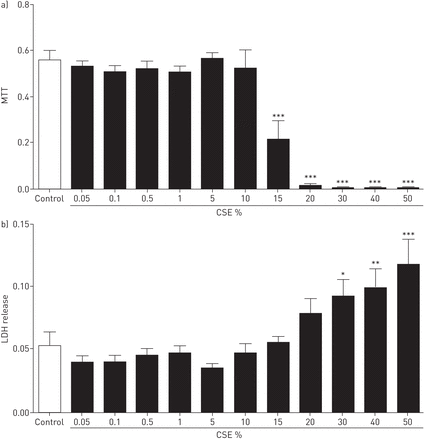
香烟烟雾提取物(CSE)对细胞活力的影响。CSE的上的细胞毒性)的线粒体活性,和b)人类气道平滑肌(ASM)细胞的乳酸脱氢酶(LDH)的释放是由Thaizolyl蓝溴化四唑(MTT)和LDH测定法在570nm / 690nm下的吸光度测定和490纳米/ 690nm处,分别。人类ASM细胞72小时(N = 4)CSE的连续稀释刺激。数据表示为平均值±扫描电镜。采用单因素方差分析+ Bonferroni后测确定统计学意义。*:p < 0.05;* *:p < 0.01;***:P <0.001,与对照组相比。
CSE诱导趋化因子
作为CSE已知诱导人ASM细胞CXCL8释放[21],我们用这样的输出来验证我们在体外模型。CXCL8释放增加10%CSE和10ng·毫升-1的TGF-β1 non-COPD和COPD组(图。图2a和b)。CXCL1的释放非COPD组中增加10%CSE和增加了COPD组中的5%和10%CSE两者;然而,TGF-β1没有引起任何一组中CXCL1的释放(图。2c和d)。两组ASM细胞间CXCL8和CXCL1的基础产量和最大产量均无差异。我们的结果显示,ASM细胞释放的CCL5和CXCL10过低,无法检测(这些方法的检测限为15.625 pg·mL)-1和31.35 PG·毫升-1, 分别)。我们发现,CCL2由ASM细胞产生,但是这不是由CSE或TGF-β1诱导,并且出现了非COPD和COPD组(数据未示出)之间没有差别。
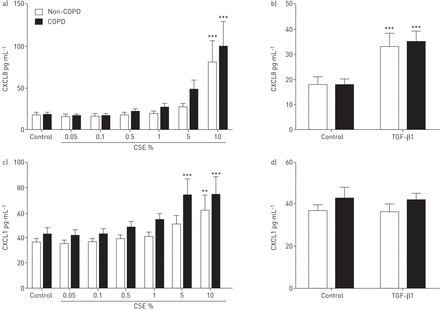
从人的气道平滑肌(ASM)细胞CXCL8和CXCL1的释放。的浓度,b)中CXCL8和c,d)在从人ASM细胞上清液CXCL1从与受试者(n = 9)和不具有(N = 9)慢性阻塞性肺病(72小时后,刺激与香烟烟雾提取物COPD)(CSE)或通过ELISA测量-β1转化生长因子(TGF)。数据表示为平均值±SEM。采用双因素方差分析加Bonferroni后测来确定统计学意义。**:P <0.01,***:P <0.001,与对照组相比。
CSE影响转录因子的DNA结合活性
我们的结果表明,无论是CSE还是TGF-β1增加转录因子的活性NF-κB(图S1a和b);然而,5% CSE仅在非copd组中增加了转录因子AP-1的活性(图S1c和d)。
ECM基因表达与粘附分子相关基因
为了研究CSE对气道重塑的潜在影响,我们使用了基于pcr的阵列作为筛选工具。70 ECM蛋白质的基因表达,粘附分子和基质金属蛋白酶是评估CSE和TGF-β1刺激(图S2)。CSE调节比TGF-β1 ECM -和粘附molecule-associated基因,还有基质金属蛋白酶基因表达差异CSE TGF-β1刺激。采用≥2分界线和/或>作为COPD和非COPD组之间的1.5倍差异,进一步研究了MMP-1、-2、-3、-10和-12、COL5、COL7和COL8A1。
CSE诱导基质金属蛋白酶
为了验证MMP的变化,我们测定MMP-1的释放,-2,-3,-10和-12从人ASM细胞。与任一CSE或TGF-β1刺激后,总MMP-12在来自人ASM细胞上清液的浓度太低而不能检测到(数据未显示)。总MMP-1的浓度的非COPD组中增加10%CSE和COPD组中增加了5%和10%CSE(图3a和图b)。无论是CSE还是TGF-β1 MMP-2影响生产(图。3c和d)。10%CSE两组增加MMP-3和MMP-10的释放,而TGF-β1COPD组中增加的MMP-3和MMP-10的释放仅(图。3E-H)。
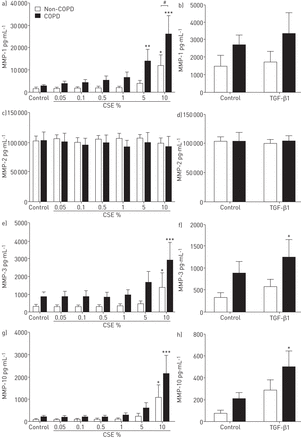
产量从人气道平滑肌(ASM)细胞基质金属蛋白酶(MMP)的。A,B的浓度)MMP-1,C,d)MMP-2,E,F)MMP-3,和G,H)MMP-10在从人ASM细胞上清液从与受试者(n = 8)和无需后72小时刺激香烟烟雾提取物(CSE)或转化生长因子(N = 7)慢性梗阻性肺病(COPD)(TGF-β1)通过ELISA测量。数据表示为平均值±SEM。采用双因素方差分析加Bonferroni后测来确定统计学意义。*:p < 0.05;* *:p < 0.01;***:P <0.001,与对照组相比。#:P <0.05,两组间的比较。
活性MMP-1的产生
为了进一步研究CSE对MMP-1的活性的影响,我们测量MMP-1的从人ASM细胞活性形式。活性MMP-1的浓度在这两个组中增加10%CSE,而TGF-β1的非COPD组中仅降低活性MMP-1的释放(图4)。
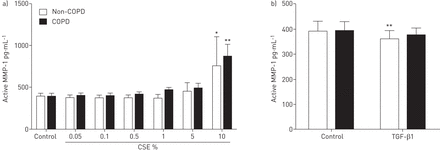
有源矩阵金属蛋白酶(MMP)的制备-1从人类气道平滑肌(ASM)细胞。活性MMP-1的从人ASM细胞上清液与浓度从受试者(n = 5)和不具有(N = 5)慢性阻塞性肺病(COPD)后72小时的刺激与香烟烟雾提取物(CSE)或转化生长因子(TGF)-β1通过ELISA测量。数据表示为平均值±SEM。采用双因素方差分析加Bonferroni后测来确定统计学意义。*:p < 0.05;**:与对照组比较,p<0.01。
ECM蛋白的沉积
验证ECM蛋白质的改变与CSE或TGF-β1刺激后,我们评估了沉积COL5, COL7和ECM COL8A1。COL7的沉积太低而无法被检测到(数据未显示)。CSE没有改变COL5的沉积(图S3)。慢性阻塞性肺病组COL8A1的沉积仅增加了0.5%、1%、5%和10%的CSE,与非慢性阻塞性肺病细胞相比,1%和5%的CSE明显增加了COL8A1的沉积(图5)。TGF-β1 COL8A1的沉积增加COPD组(图5 b)。CSE没有从任一基团,其是在成纤维细胞中[形成鲜明对比的先前的发现改变纤连蛋白的沉积12, CSE抑制两组人的ASM细胞中perlecan的沉积(图S4)。
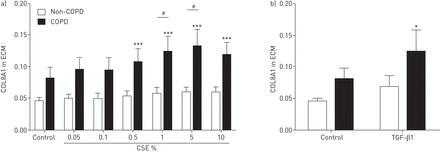
胶原VIIIα1(COL8A1)来自人类气道平滑肌(ASM)细胞的沉积。在从人ASM细胞从与受试者的细胞外基质(ECM)的沉积COL8A1(N = 7)和不具有(N = 7)慢性梗阻性肺病(COPD)后72小时的刺激与香烟烟雾提取物(CSE)或转化生长因子(TGF)-β1通过ECM ELISA在450nm / 570nm处的吸光度进行测定。数据表示为平均值±SEM。采用双因素方差分析加Bonferroni后测来确定统计学意义。*:p < 0.05;***:P <0.001,与对照组相比。#:P <0.05,两组间的比较。
细胞外基质蛋白的释放
为了验证与CSE或TGF-β1刺激后的可溶性细胞外基质蛋白的改变,我们评估使用免疫印迹上清可溶性COL1和可溶性纤维连接蛋白。我们的研究结果表明,无论是CSE也不TGF-β1显著上升COL1的释放(图S5)。我们的研究结果还显示,CSE没有引起可溶性纤维连接蛋白的释放;然而,TGF-β1显著增加纤连蛋白的释放(图S6)。
COL8A1呼吸道支气管
正如我们在COPD ASM细胞中发现更大的CSE诱导COL8A1,我们接下来的调查,如果我们在体外结果反映了慢性阻塞性肺病在活的有机体内。免疫组化显示COL8A1在COPD患者(n=10)和COPD患者(n=7)的气道组织中均有表达。在两组气道中,COL8A1似乎定位于基底膜、血管壁和ASM束。阳性染色采用阈值和同型对照染色。通过密度分析(染色面积的量化),我们发现COPD组COL8A1的整体表达更高(图。6)。

胶原蛋白VIII - 1 (COL8A1)气道组织染色。a)免疫组化检测10例(n=10)和7例(n=7)慢性阻塞性肺疾病(COPD)患者气道支气管COL8A1。用化学染色团DAB(棕色)对细胞核进行特异性染色,苏木精(蓝色)对细胞核进行反染。苏木精和伊红染色观察组织结构。酒吧= 100μm规模。b)非COPD组及COPD组COL8A1免疫染色以阳性染色面积量化,并以同型对照纠正。数据以中位数表示。采用Mann-Whitney检验确定统计学意义。*:p < 0.05。
CSE抑制细胞附着和伤口愈合
为了进一步研究CSE对慢性阻塞性肺病患者(n=5)和非慢性阻塞性肺病患者(n=5)的ASM细胞功能的影响,我们评估了慢性阻塞性肺病患者(n=5)和非慢性阻塞性肺病患者(n=5)的ASM细胞的细胞附着情况。CSE显著降低了COPD组的ASM细胞对培养板的附着(图。7)。伤口愈合分析表明CSE的高浓度在人类ASM细胞(n = 6)(图S7)显著降低伤口愈合的速度。
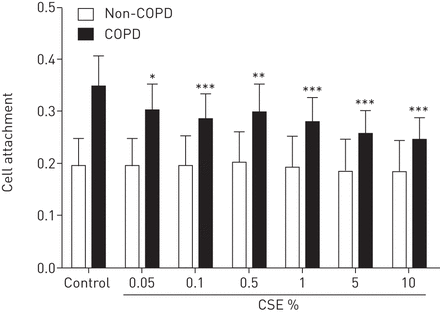
上2小时的细胞附着香烟烟雾提取物(CSE)的在595nm的吸光度的影响。对人呼吸道的CSE处理板细胞附着平滑从受试者肌肉细胞(N = 5)和不具有(N = 5)慢性阻塞性肺病(COPD)通过甲苯胺蓝测定法测量。数据表示为平均值±SEM。采用双因素方差分析加Bonferroni后测来确定统计学意义。*:p < 0.05;* *:p < 0.01;***:P <0.001,与对照组相比。
讨论
我们从与不吸烟者慢性阻塞性肺病发现不同的反应,以CSE在ASM细胞。具体地,MMP-1和COL8A1的ASM中细胞中的沉积分别增加了CSE,而这些增加是COPD组中更高。我们的结果还表明,CSE降低ASM细胞附着到培养板和卷绕从患有COPD的吸烟者中分离的细胞特异性愈合。这些结果表明吸烟者认为ASM细胞与COPD是CSE刺激可以部分解释,慢性阻塞性肺病的发展在某些吸烟者更敏感。
慢性阻塞性肺病是一种炎症性疾病,其特征是中性粒细胞数量增加[9],[22],以及在支气管肺泡灌洗液中增加中性粒细胞趋化因子(如CXCL8和CXCL1)的数量[23],[24]。CSE是CXCL8在ASM细胞的有效诱导剂[21],[25]。然而,CXCL8的高分泌是否在从COPD患者中分离如在其它气道细胞ASM细胞发生是未知的[26],[27]。因此,我们还测量了CSE诱导的CXCL8,发现CSE增加CXCL8从ASM细胞释放还没有从人与不COPD细胞之间的差异。我们还测量了CSE诱导CXCL1,发现CSE从ASM细胞增多CXCL1的释放。此外,来自COPD患者ASM细胞对刺激CSE用于生产CXCL1的更敏感。无论CXCL8和CXCL1有相似的生物学特性,因为它们都对嗜中性粒细胞的招聘[影响28] - [30.]。在我们的研究中,我们使用了患有和未患有COPD的吸烟者的ASM细胞。CSE在两组ASM细胞中诱导CXCL8和CXCL1可能反映了COPD患者中性粒细胞增多的观察[31]和吸烟无COPD [32]。此外,由于低浓度的CSE仅在COPD细胞诱导CXCL1,这表明这些细胞是高反应性,以CSE,或许可以解释为什么有些吸烟者制定COPD和别人不一样。
我们的结果表明,高浓度的CSE的增加既前-MMP-1和活性MMP-1(间质胶原酶)的来自人ASM细胞释放,并且这似乎与COPD比没有COPD的吸烟者更明显。当我们仅测量活性MMP-1,我们发现从两组细胞之间的类似的生产。这表明在该ASM产生活性MMP-1的可能不COPD中肺病理学的关键决定因素,但可能与工艺中常见到具有和不COPD两者吸烟者。在其它细胞中,CSE增加了生产MMP-1的从其中似乎通过细胞外信号调节激酶-1/2促分裂原活化蛋白激酶途径主要驱动人上皮细胞和人肺成纤维细胞[33],[34]。我们还发现,人的ASM细胞组成性地产生了高水平的MMP-2(明胶酶A),而CSE并没有增加MMP-2,这与暴露在香烟中的成纤维细胞的发现形成了对比[35]。
与其他基质金属蛋白酶相比,基质金属蛋白酶-3 (stromelysin 1)和-10 (stromelysin 2)迄今未被广泛研究。我们的研究表明,CSE增加了人ASM细胞中MMP-3和-10的基因表达,10% CSE增加了MMP-3和-10的产生。这些结果表明,MMP-3和-10可能导致慢性阻塞性肺病吸烟者不同的进展。两项基因分型研究表明,MMP-3多态性与COPD疾病进展相关[36],[37]。另一项研究显示,MMP-10基因在小气道和小气道周围软组织中的表达均增加,与COPD进展相关[38]。此外,我们的结果发现MMP-12(巨噬细胞弹性酶)基因表达通过CSE增加在体外。然而,MMP-12蛋白的从CSE释放刺激ASM比测定(9.2皮克·毫升的检测极限下-1)。
此前已有研究表明,CSE增加了COPD成纤维细胞中纤连蛋白和perlecan的沉积[12],所以在这项研究中,我们还通过ASM细胞中,测量他们的生产。然而,CSE不影响生产的COPD ASM细胞纤维连接蛋白,降低生产的串珠,表明对CSE反应是细胞类型特异性的。我们的阵列数据显示,几个胶原响应CSE刺激也发生了变化,所以我们选择评估COL5,COL7和COL8。COL5的蛋白水平响应CSE没有改变,并且这些工具来衡量COL7是不可靠的。然而,CSE诱导从COPD比从控制单元的ASM更大COL8生产。很少有知道关于COL8在慢性阻塞性肺病,尤其是不COL8的人与不COPD气道的量。在这项研究中,我们发现,COL8的表达在COPD气道增加,特别是在和周围的平滑肌束,这表明平滑肌产生COL8原位并且指示COL8的从吸烟者患有COPD的沉积是可能有助于气道病理学。COL8具有短的三螺旋并且含有α1和α2链,并且每一个α链包含胶原结构域,短的N末端的非三螺旋区域(NC2)和较长的C-末端非三重螺旋结构域(NC1)。如已知的其它胶原,COL8的不同区域可以具有相反的生物效应。例如,整个COL8分子增加主动脉平滑肌细胞增殖和迁移[39],同时COL8A1的NC1区抑制牛主动脉内皮细胞的线粒体活性40]。COL8以前没有被报道为COPD ECM的决定因素,但是暴露在污染环境中的怀孕小鼠导致其后代肾小管细胞中COL8的增加[41]。这就提出了一个问题,是否会发生在谁吸烟孕妇的后代相似的遗传效应。此外,这种风险的功能和长期的结果没有被检查。
如果MMP-1和COL8之间的相互作用发生,我们没有调查,就再也没能在文献中找到具体的例子。很容易将其推测增加的MMP能够降解胶原;然而,我们发现,它们都在相同的时间的增加,显示出的净效果是胶原蛋白沉积。
慢性阻塞性肺病患者的小气道壁和肺泡壁的修复受损[6],[7]。我们的研究结果表明,CSE仅减少慢性阻塞性肺病患者的ASM细胞的细胞附着,而高浓度的CSE也会降低ASM细胞的伤口愈合率。这些结果可能表明,与慢性阻塞性肺病吸烟者和非吸烟者相比,ASM细胞存在先天差异。一项关于CSE对人肺功能影响的研究表明,CSE降低了正常人支气管平滑肌细胞的迁移和收缩活性[42]。在另一项研究已表明CSE损害伤口牛支气管上皮细胞的愈合通过活性氧物质依赖的机制[43]。另一项研究显示,CSE可抑制人肺成纤维细胞的增殖[44,但这些成纤维细胞是否来源于COPD患者尚不清楚。
调查如果转录因子活性的差异可以解释增加的响应性的慢性阻塞性肺病ASM细胞CSE我们选择测量转录因子的活动NF-κB AP-1,这些曾被证明参与从ASM细胞基质金属蛋白酶和细胞因子的释放45],[46]。我们没有发现COPD ASM细胞中转录因子活性增加的证据。这表明,慢性阻塞性肺病细胞对香烟烟雾的反应增强可能是由于表观遗传变化,正如我们在慢性阻塞性肺病的其他细胞中所发现的那样[47]。
综上所述,我们发现在吸烟刺激下,慢性阻塞性肺病患者和非慢性阻塞性肺病患者的ASM细胞产生的趋化因子、基质金属蛋白酶和胶原蛋白存在差异。从慢性阻塞性肺病患者中分离出来的ASM细胞在释放炎症介质和与气道重塑和细胞行为相关的因子方面对香烟烟雾有更高的反应。我们从这份手稿中获得的数据表明,慢性阻塞性肺病中的ASM能够对香烟烟雾中的可溶性成分作出反应。这将通过(释放趋化因子)募集中性粒细胞,并可能通过改变ECM的沉积影响其他气道细胞。由于平滑肌数量与COPD严重程度呈正相关,这些影响可能在重度COPD中被放大。因此,我们的研究结果提示COPD吸烟者的ASM细胞参与了该病的病理发展。
致谢
w ^e would like to acknowledge the collaborative effort of the cardiopulmonary transplant team and the pathologists at St Vincent’s Hospital (Sydney, Australia), and the thoracic physicians and pathologists at the Royal Prince Alfred Hospital (Sydney) and Strathfield Private Hospital (Strathfield, Australia).
脚注
这篇文章有补充资料www.qdcxjkg.com
利益冲突:可以在www.qdcxjkg.com上找到这篇文章的网络版
- 收到2013年10月1日。
- 公认2014年5月4日。
- ©2014人队











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}