





文摘
在肺内的肾素-血管紧张素系统通过组织血管紧张素ⅱ的浓度或缓激肽可能有多个影响肺部病理生理学。因此,研究是否D等位基因的存在的血管紧张素转换酶(王牌)插入/缺失(I / D)多态性或血管紧张肽原的等位基因(AGT)启动子多态性(6)30天/ G是独立的风险因素在急性呼吸窘迫综合征(ARDS)患者生存。
在未来的研究中,成年人(德国人白种人的种族)与ARDS (n = 84)招募从目前作者的重症监护室和基因分型王牌I / D和AGT(6)A / G多态性,200名健康白种人的控制。
死亡率增加王牌DD基因型与我相比等位基因,王牌I / D多态性是一个30天的生存的独立预后因素。一个纯合子患者DD基因型在最高的死亡风险(风险比5.7;95%置信区间1.7 - -19.2)与II基因型。相比之下,AGT(6)A / G多态性既不是为ARDS的发展与风险增加有关也与结果。
在急性呼吸窘迫综合征患者中,血管紧张素转换酶插入/缺失多态性而非血管紧张肽原(6)A / G启动子多态性是一个独立的危险因素对30天的生存有着明显的影响。
急性呼吸窘迫综合征(ARDS)的特点是增加capillary-alveolar渗透率、低氧血,减少肺合规、双肺浸润扩散和机械通风的必要性。ARDS仍然是一个重要的死亡原因在重症监护室(ICU)和一些特定的治疗方法是可用的。因素预测这种综合症的发病或严重性并不广为人知,但ARDS的发病率低风险相对较大的组患者中表明遗传和环境因素之间的相互作用1,2。ARDS的早期阶段的特点是高渗透率肺部水肿,肺泡上皮细胞和嗜中性粒细胞浸润,损失可能进步显著肺泡间质改造和纤维化。肺的实验证据表明,激活肾素-血管紧张素系统(RAS)可能会影响ARDS的发病机理通过机制影响血管通透性3、血管的语气4,纤维母细胞增殖5和减少肺泡上皮细胞的生存6。
因此血管紧张素(AT)——ARDS的发病机制中可能发挥重要作用。AT-II是通过蛋白水解生成的正常人血管紧张素原(AGT)解理,主要是在肝脏合成,在较小程度上,在肾、脑、心脏、肾上腺的键盘,脂肪和血管壁7,8。AGT首先转换通过肾素十肽在我。此后,血管紧张素转换酶(ACE)将在我AT-II。然而,高手也降解缓激肽,对血管张力的影响,血管渗透性和心脏功能9。因此,血管舒缓激肽也可能导致ARDS的发病机制。
大约47%的方差在血浆ACE活动解释为常见的插入/缺失(I / D)多态性位于内含子的16王牌基因,D等位基因与ACE活性增加有关10。循环血管往往降低ARDS患者11。然而,这可能反映了减毒酶释放受损的肺血管内皮和可能不代表其他肺ACE活动隔间。事实上,增加王牌活动报道支气管肺泡灌洗(BAL)液体,尽管循环浓度下降11。符合这一观察,AT-II transpulmonary梯度和循环浓度的提高ARDS患者12和循环RAS激活,transpulmonary梯度的保护,还发现在危重病人13。当王牌主要来源于内皮细胞14和肺是人体最大的内皮表面,个人间的差异王牌活动可能影响的敏感性,和生存期间,ARDS15。自从AGT血浆浓度接近酶反应的米氏常数肾素和AGT之间血浆AGT水平的上升会导致增加并行AT-II形成16。
人类的AGT港口(6)a / G基因启动子多态性17。(6)等位基因显示启动子活动增加与(6)G相比,与此同时增加等离子体AGT的水平18。因此,如果AT-II ARDS的发病机理是至关重要的,基因型的AGT (6) A / G多态性也可能影响的敏感性,和生存期间,ARDS。
因此,研究基因型的王牌I / D多态性或AGT(6)A / G启动子多态性是独立的风险因素30天ARDS患者的生存期。
方法
研究人群
本研究回顾和大学医院的伦理委员会批准的埃森(德国埃森)。在一段时间内2年,大学专业ICU住院的病人埃森研究被认为是合格的,如果他们完成了联合美国/欧洲共识委员会ARDS的标准19,没有先前的历史ARDS和书面知情同意。所有的病人(43岁男性,41岁女性;平均年龄43±16岁)是白人种族的德国人。特定的临床疾病诱发ARDS决心在病人见面后第一个24 - 48 h ARDS的标准。在其他的研究中,一些病人有超过一个潜在诱发临床状况与ARDS的发展有关20.。然而,负责的主要条件是由调查人员经过仔细审查的临床和实验室数据。主要的临床疾病包括肺炎、败血症和其他障碍(如。创伤,愿望)。肺炎被定义为主要的证据肺部感染由细菌,病毒,赋格曲的或寄生虫感染诊断的医学历史、革兰氏染色和文化气管吸入或落下帷幕的标本。脓毒症综合征这个词被用来定义的骨头20.。患者脓毒症综合征和肺炎都满足标准分为肺炎、肺炎诱发败血症的假设被综合症。在基线临床和人口数据,包括肺损伤评分(LIS),简化急性生理学得分(SAPS)21和连续的器官衰竭评估(沙发)得分22,计算病人见面后第一个24 - 48 h ARDS的标准。病人被归类为幸存者,如果他们还活着后30天ARDS的诊断或从医院出院没有机械通风的要求。
控制样本包括200名健康的高加索人的输血医学、招募当地部门埃森大学医院。所有随机样本收集受试者献血和样品的细节已经公布23。
确定基因型
DNA被修改phenol-chloroform从全血中提取样本提取。王牌基因型是由剪接位PCR扩增如前所述24对所有主题数据,由工作人员不清楚。这种方法收益放大产品D等位基因的84个基点和65个基点的等位基因,和聚丙烯酰胺凝胶产品视觉效果。纯合子插入(II)基因型重复PCR证实了在缺乏我等位基因的引物。所有样品都是随机,由两个独立的研究人员和结果的基因复制。基因分型的AGT(6)A / G多态性,使用引物进行PCR AGT-Pro-SE 5′-CTTCTGGCATCTGTCCTTCTGG-3′生物素化的和引物5′AGT-Prom-AS -CCTAGCCCACAGCTCAGTTACATC-3′,导致200个基点片段。基因型测定使用焦磷酸测序25与测序引物5′AGT-Pro-Seq -GGCAGCTTCTTCCCC-3′。
统计分析
本研究的临床结果分析是依赖生存的第一个30天王牌和AGT基因型。使用kaplan - meier情节和生存率较趋势评估日期的基因型和临床结果之间的关系的主要诊断的后续。Log-rank测试王牌基因型进行使用所有三个基因型,除非另有规定。电源(1-β)的临床结果分析是0.87。性的影响,削弱了、沙发、LIS、年龄、身体质量指数(BMI)、呼气末正压通气(偷看),动脉氧张力和吸入氧气的比例分数(P啊,一个2/F阿,我2),王牌基因型为临床预后因素逐步多元Cox回归分析的结果进行了分析。无意义的变量(p > 0.05)逐步从模型中删除。风险比率(人力资源)和95%置信区间(CI)计算Cox回归模型包括所有剩余因素的多变量分析和单变量分析表明因素。应急表和皮尔逊卡方测试被用于分类变量使用王牌和AGT基因型。自王牌I / D多态性似乎显示gene-dose效应,连续参数方差分析被用于比较变量,在适当的地方。克鲁斯卡尔-沃利斯检验是用于连续非参数变量。哈迪温伯格平衡被拟合优度检验卡方测试。差异,p值< 0.05被认为是重要的。给出了连续变量均值±sd,如表示。
结果
使用kaplan meier估计比较结果,30天的存活率显著相关王牌基因型(p = 0.011;图1⇓)和一个明显gene-dose效果。病人的纯合子王牌DD基因型显示患者的死亡风险显著高于II基因型,杂合的患者在中间风险(人力资源:弟弟与2:3.6 (95% CI 1.3 - -8.7), p = 0.011;ID与2:2.2 (0.7 - -6.1),p = 0.162)。术后存活率为73.2%二世,DD基因型,分别ID为64.0%和50.0%(图。1⇓)。相比之下,没有这样的协会发现AGT基因型(图2所示⇓)。
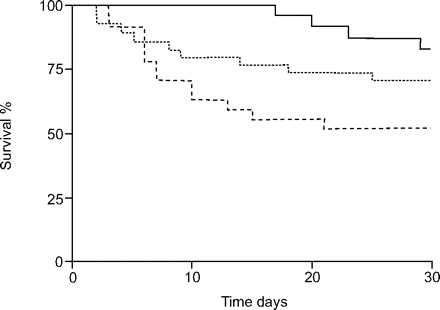
急性呼吸窘迫综合征患者的术后生存依赖的血管紧张素转换酶基因插入/缺失(I / D)多态性。用来计算概率的kaplan - meier估计30天的生存,在II基因型相比显著增加与运营商的D等位基因(p = 0.011)。- - -:2;▒:身份证;- - -:弟弟。
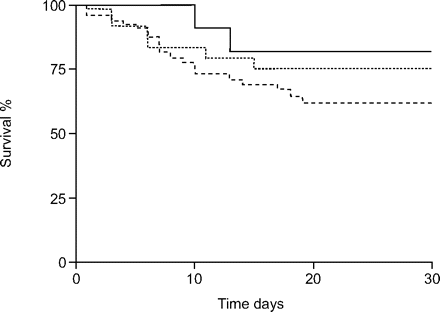
急性呼吸窘迫综合征患者的术后生存依赖于血管紧张肽原基因的启动子多态性。用来计算概率的kaplan - meier估计30天的生存。术后生存率没有明显不同血管紧张素启动子多态性基因型(p = 0.28)。- - -:AA;▒:GG;——————:GA。
多元比例风险分析包括年龄、性别、LIS、BMI,偷看,削弱了P啊,一个2/F阿,我2不显示的王牌I / D多态性是最重要和30天的生存的独立预后因素。纯合子的DD受试者的死亡风险最高(HR 5.7 (95% CI 1.7 - -19.2), p = 0.005)和II基因型相比(表1所示⇓)。
临床ARDS患者的描述王牌I / D基因型是显示在表2⇓。无显著关联的王牌基因型被发现与性别、年龄、LIS、BMI, P啊,一个2/ F阿,我2偷看,削弱了,沙发或ICU停留时间。同样,这些变量是相关的AGT基因型(表3⇓)。
ARDS的最常见原因是肺炎(67%)、脓毒症(24%)、外伤(4%)和愿望(5%),和总体生存率是68%。人口特征的ARDS患者和健康对照组显示在表4中⇓。关于人口特征之间没有明显的统计学差异ARDS和对照组被发现,除了年龄的细微差别。平均年龄是43±16岁ARDS组和对照组37±2岁。
两个样本,基因型分布与哈迪温伯格平衡相一致。王牌和AGT基因型和等位基因频率ARDS患者和献血者样本之间的相似(表4所示⇑)。王牌和AGT基因型没有在连锁不平衡,因此没有相互作用王牌和AGT基因型和一个观察与ARDS。
讨论
目前的研究显示第一次的D等位基因的多变量分析王牌I / D多态性显著增加30天死亡率ARDS患者。近6的人力资源为纯合子的DD和∼5杂合的ID基因型与II基因型相比,不仅表明,D等位基因可能对肺内的ACE的活动一个重要的影响系统,但也强调的潜在相关性肺ACE系统病理变化。D等位基因之间的联系和发展或结节病的进展26、哮喘27和铍中毒28已被描述。由于肺部RAS对血管通透性的影响3、血管的语气4,纤维母细胞活动5和肺泡上皮细胞的生存6ARDS的自然过程,其很强的影响是可以理解的。在肺中,肺循环是一个重要的潜在的RAS激活。血管紧张素转换酶抑制剂在健康人体内减弱肺血管收缩和肺心病患者4,1型受体拮抗剂29日。注入在我30.或AT-II3可以唤起性肺水肿独立于儿茶酚胺的释放。因此,AT-II可能也影响微血管通透性。最后,AT-II是肺泡上皮细胞pro-apoptotic因素在体外31日。失去了一个完整的上皮屏障(影响血管之间的流体的运动和细胞,间质和肺泡空间)是另一个ARDS的早期事件,可能会受到RAS。任何单一的机制或所有机制相结合可能对ARDS及其结果的影响。
观察一个重要协会D等位基因和死亡率之间的患者纳入本研究。几个途径影响ARDS和/或额外的机制可能涉及。例如,fibroproliferation可以在ARDS的结果有显著的影响5,32它曾表明,AT-II肺成纤维细胞的有丝分裂原5。此外,血管紧张素转换酶抑制剂和1型受体拮抗剂减弱胶原沉积和间质纤维化肺损伤的实验模型33。患者的左心室功能障碍,ACE抑制改善气体传输和ventilation-perfusion-coupling至今未知的机制34。因此,它会不会意外如果王牌活动增加,相关的王牌D等位基因,ARDS患者诱发相反的影响。最后,ACE表达激活肺泡巨噬细胞35和淋巴细胞10。在肺巨噬细胞激活,ACE抑制减少自由基的表情;然而,它的作用在调节炎症反应并没有明确定义35。
众所周知,I / D多态性占47%的方差在血浆ACE活动健康白色个人,ACE活性最高的那些DD基因型10。然而,ACE浓度和活动不仅受基因型的影响,也被无数的机制,包括药物治疗36和液体管理37。因此,当前作者没有试图测量浓度或在BAL液体或血清ACE活性。连接和分离分析表明,循环ACE水平影响主要数量性状位点地图内或接近王牌基因。多个变种,在连锁不平衡I / D多态性描述,但这些变异与ACE水平有关。此外,它不能被排除在外的王牌在连锁不平衡I / D多态性与一个或多个在以外的基因变异王牌这些未知的单核苷酸多态性解释这里描述的关联38,39。即使多的信息表明AT-II ARDS的主要因素,它还应该记住ACE降解缓激肽,也有对血管张力的影响,血管渗透性和心脏功能9。
出乎意料,AGT(6)A / G多态性在ARDS并不影响生存,尽管基因型多态性可能与血浆AGT浓度的增加,随后增加AT-II一代16,18。因此,目前的结果可以解释的假说的协会看到王牌I / D多态性与缓激肽降解而不是增加AT-II形成。
的基因型之间的联系王牌ARDS I / D多态性和敏感性,马歇尔的建议et al。15,无法证实在高加索示例研究目前,尽管患者群体仅略有不同临床特点,如削弱了LIS),P啊,一个2/F阿,我2、ICU停留时间和整体死亡率。然而,一个重要的区别可能各自ARDS患者的病因学。在目前的研究中,肺炎是ARDS的根本原因在60%的患者中,而只有30%的病例被诊断为肺炎马歇尔的样本et al。15。因此,可以推测ARDS的D等位基因和易感性之间的关系只存在于二次(nonpneumonia) ARDS。这个假设似乎是由生成的数据关于亚洲流行的严重急性呼吸系统综合症(SARS),作为ARDS的易感性的D等位基因无关王牌多态性40。
尽管30年的研究机制和ARDS的后果,努力确定一个可靠,pulmonary-specific死亡危险因素一直令人失望。独立变量与死亡率相关肺部病理生理学的不是具体的异常,如败血症、nonpulmonary器官系统功能障碍,年龄或肝硬化41,42。尽管指数低氧血,等P啊,一个2,F阿,我2或者是P啊,一个2/F阿,我2比,最初被认为具有预后价值42,43,后续研究证实这些变量没有独立与死亡的风险当他们测量了ARDS的早期过程中41,42。此外,大多数ARDS患者最终死于多器官功能衰竭和目前的研究不能解释为什么王牌基因多态性影响的结果。此外,当前作者不知道另一个路径依赖的基因型王牌多态性被触发。例如,有实验室证据表明血管紧张素参与核factor-κB的激活。这可能表明促炎效应发展或恶化的全身炎症反应综合征44。然而,这些只是猜测。此外,ARDS,根据定义,包含一个相对较小的范围内表型的肺功能障碍,几乎所有的患者表现出严重的功能障碍,肺动脉高压和高分路。因此,这将是意想不到的表型的相关性(分流器等)和基因型被发现。
尽管所有ARDS患者接受一个标准化的多通道的概念包括analgosedation、液体,通风,血液动力学的,抗生素和诊断管理,它不能被排除在外,由于障碍的多因子的性质,未知的,潜在的混杂因素仍然存在。因此,本研究的结果不应过分的解读。然而,目前的调查显示王牌I / D多态性影响肺疾病,也凸显了ARDS的ACE系统的潜在相关性。
总之,目前的研究可能会在未来的临床影响。这种影响可能包括识别缓激肽的新型候选基因途径,调整相关的药物治疗的肾素-血管紧张素系统和分层病人进行临床试验。
确认
作者要感谢美国Costabel(德国埃森)的批判阅读手稿。
- 收到了2006年4月3日。
- 接受2006年11月3日。
- ©人期刊有限公司











{kind=link}
{kind=link}
{kind=link}
{kind=link}