





摘要
淋巴水肿,骨髓增生异常,纤维化和肺泡蛋白沉积GATA2突变http://ow.ly/RHci303h8ks
编辑器:
肺泡蛋白沉积症(Pulmonary alveolar proteinosis, PAP)是一种罕见的综合征,其特征是表面活性蛋白在肺泡内异常积聚,从而损害气体交换[1].PAP被分为三组:自身免疫杂志(APAP),由抗粒细胞 - 巨噬细胞菌落刺激因子(GM-CSF)自身抗体,继发性PAP(SPAP)和遗传杂库定义。血液学疾病是SPAP的最常见原因,特别是骨髓增生术综合征(MDS)[1,2].PAP的诊断基于胸部计算机断层扫描(CT),该扫描显示疯狂的铺路模式,以及支气管肺泡灌洗液(BAL)的典型高碘酸-希夫(PAS)染色阳性[3.].目前,很少需要手术肺活检来诊断PAP,除非出现不典型的表现,如以下所述的病例,伴有GATA结合蛋白2 (GATA binding protein 2)缺乏的患者并发肺纤维化和sPAP。
2013年11月,随着纽约心脏协会(NYHA)的增加,一名来自北非的28岁白人女性,不吸烟者,一家燃料公司的高管,住进了Pontchaillou医院(法国雷恩)的呼吸科2期呼吸困难、干咳和间质性肺病。除诊断为右下肢特发性淋巴水肿外,她没有明显的家族史和既往病史 几年前(图1一个).临床检查发现指状突,胸部听诊正常。胸片显示双肺尖处可见可伸缩网状混浊。胸部CT显示双侧小叶间及小叶间隔弥漫性增厚,以上叶为主,并伴有丰富的胸膜下网状结构(图1 b)肺活量测定显示出一种纯粹的限制模式:总肺活量为预测值的68%,强迫肺活量(FVC)为预测值的52%。一氧化碳的肺转移因子(TLCO)减少(40%的预测值)。BAL呈乳白色,发现含有50 000 细胞·mL−1巨噬细胞占72%,淋巴细胞占26%。PAS染色无法在BAL上进行,手术肺活检已完成。组织学检查显示内脏胸膜明显增厚,胸膜下和膈旁纤维化明显。间质纤维化伴随着充满嗜酸性蛋白物质的肺泡,这种物质被证明是PAS阳性和抗淀粉酶的。空泡状、泡沫状的肺泡巨噬细胞和胆固醇晶体也很常见。罕见的成纤维细胞病灶和轻度慢性炎症。蜂窝状、淋巴聚集、细支气管化、平滑肌增生、鳞状化生、纤维弹力增生和肉芽肿均不存在(图1 c).血清中未发现抗gm - csf抗体。血液学sPAP怀疑是由于血细胞减少:血红蛋白10 g·dL−1,血小板50 g·L−1,白细胞3.9 g·L−1中性粒细胞3.34 克·升−1,嗜酸性粒细胞0.04 g·L−1,嗜碱性粒细胞0.15 g·l−1,淋巴细胞0.33 g·L−1和单核细胞0.04 g·l−1淋巴细胞表型显示B淋巴细胞、T淋巴细胞和自然杀伤淋巴细胞减少。丙种球蛋白正常:12.4 克·升−1.骨髓活检证实MDS(多系发育不良和<5%核型正常的母细胞).由于患者的年轻、淋巴水肿、MDS和sPAP之间存在关联,因此寻找GATA2缺陷并呈阳性:外显子5 C.1020_1029dup:p.R344GfsX43。在9个月的时间里,MDS保持稳定,尽管她的呼吸困难症状明显恶化,达到NYHA阶段3呼吸困难,肺功能下降ts(FVC从52%降至40%和TLCO从预测值的40%到25%下降)。然而,她的胸部射线照片保持不变。2014年9月进行双侧全肺灌洗,对她的呼吸道症状,肺活量测定或胸部CT扫描没有任何影响。这导致我们考虑血液吞咽干细胞移植(HSCT)。她的兄弟是HLA匹配的,GATA2缺乏是消极的。
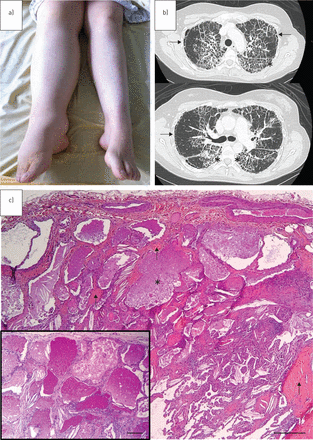
a)右肢体淋巴水肿。b)胸部计算的断层扫描图像,显示在下叶(星号)的上裂片和顶部叶片和顶部叶片中的弥散性,双侧和不规则增厚,显然是主要的叶片(星号)。在上叶的沉窗区域中,我们观察了探针肺气肿(箭头)。c)手术肺活检。注意肺泡空间内的嗜酸性,粒状,蛋白质材料,肺肺泡蛋白病变(星号)。该材料是定期的酸 - 席夫(PAS) - 阳性(INSET)。相关的间质纤维化由箭头显示。Haematoxylin-eosin-Safran:秤杆=500μm;PAS(插图):秤杆=250μm。
据我们所知,这是一种新的框架转换GATA2在诊断时GATA2缺陷时,突变和严重间质性肺纤维化伴PAP的罕见描述之一[4].这可能是进行性慢性PAP的最终结果,也可能是GATA2缺乏症患者PAP的特异性结果。米archand -一个坝et al。[5]描述了一名35岁男性的患者,具有特发性骨髓性发作性骨髓性骨髓Aplasia,随后随后通过与杂合子有害种类相关的特发性肺纤维化叔(端粒酶逆转录酶)突变。同样,一名63岁的女性诊断出aPAP, 7年后演变为肺纤维化[6].弥漫性纤维化改变取代了疯狂铺路型,呈现胸膜下蜂窝型。这一发现可能提示肺纤维化的发病机制中存在表面活性物质功能障碍[6].在文献中,SPAP的诊断,管理和预后不同于其他类型的PAP。胸部CT扫描的演示文稿更为非典型,垫的预后比APAP更差(2年生存率为46%与100%) [2,3.,7,8].SPAP的管理通常基于潜在疾病的治疗方法[1,2].化疗和早期造血干细胞移植可治疗sPAP。全肺灌洗亦是改善病人症状的有效治疗方法[1,2,4].
在本病例报告中,即使在罕见肺病能力中心,肺科医生和放射科医生一开始也没有考虑PAP诊断。Pontchaillou医院呼吸科在处理PAP方面经验丰富,在过去的15年里对PAP患者进行了60多例全肺灌洗[1,9].之后不能对BAL进行PAS染色。经支气管活检,特别是冷冻活检可能是肺间质性疾病的可靠诊断工具。然而,鉴于这种不典型的胸部CT表现,血小板减少导致我们进行手术肺活检,这是一种更容易出血的单一风险。的确,上叶间隔增厚与疯狂铺路型相吻合,并伴有丰富的胸膜下网状结构导致纤维化,这是出乎意料的发现。与aPAP相比,sPAP更难诊断,这不仅是因为胸部CT表现不典型,也因为缺乏抗gm - csf抗体,可能存在潜在疾病。与aPAP相比,sPAP的表现更分散,地域分布更少:62%与19%和24%与分别为71% (8].sPAP患者胸膜下毛玻璃样混浊和铺路样病变的发生率明显降低[8].手术肺活检的使用比APAP更频繁(41%)与7%)[7].因此,通过不明确的间质肺病疾病,PAS染色是对BAL进行的有趣测试,以避免手术肺活检。
在I什叶派et al。[2] 31例伴MDS的sPAP患者中,PAP进展是导致死亡的主要原因。感染和MDS的进展分别是死亡的第二大和第三大原因[2].31例MDS患者中只有10例接受全肺灌洗治疗,只有3例对治疗有积极反应[2].
GATA2属于锌指转录因子家族,是造血细胞中基因表达的关键调节因子,最近发现GATA2缺乏[10].GATA2已被证明可以调节肺泡巨噬细胞吞噬作用。在GATA2缺乏症中,鉴于受PAP影响的患者的BAL流体中的肺泡巨噬细胞的丰度,存在肺泡巨噬细胞功能障碍,而不是定量缺陷[10].
年代平纳et al。[10]描述了57例经证实的导致GATA2缺陷的突变患者的临床特征。他们发现了一种广泛的表型,包括免疫缺陷、血液病、PAP和淋巴功能障碍,这与我们的患者相符。54%为女性,初次就诊时中位(范围)年龄为30(4-76)岁,初次就诊时中位(范围)年龄为20岁(5个月至78岁)。在接受骨髓活检的50名患者中,42名患者(84%)符合MDS的诊断标准,大多数骨髓活检显示多系发育不良和小于5%的原始细胞。6例患者(11%)存在慢性淋巴水肿。活检证实的PAP在10名成人中被发现(18%)[10].在S的讨论部分平纳et al。[10,作者报道,在他们的队列中扩散和通气缺损的高患病率可能反映了复发性肺部感染继发的肺泡充盈、肺气肿改变、纤维化或支气管扩张。然而,并没有具体描述伴随PAP的肺纤维化。20岁时的总生存率为96%,40岁时为77%,60岁时为45% [10].21例患者接受了MDS,PAP或反复感染的HSCT。所有患者患者患有肺功能的显着改善[10,11].重要的是,家庭成员应定期接受筛查GATA2在捐献骨髓之前发生突变在本病例报告中,现在考虑HSCT。我们期待在同种异体移植前进行新的肺部评估。正在与患者、她的家人、肺科医生和目前的移植团队进行讨论,评估风险/收益平衡。事实上,死亡风险随着TLCO或用力呼气量为1 s≤65%的预测,NYHA 4期呼吸困难或氧疗[1].此外,sPAP生理病理学支持肺移植后复发,因为肺泡巨噬细胞在定量和定性上无法提供表面活性剂清除[4,8].
总之,这是伴随着严重肺纤维化和诊断的概念性结缔组织在诊断中复杂化的罕见描述之一GATA2突变。这一非常罕见的综合征对诊断很重要,因为在呼吸状况恶化之前讨论了HSCT。
致谢
作者感谢Marie de Tayrac(法国雷恩大学雷恩1号génétique moléculaire et génomique服务中心,hôpital Pontchaillou和UMR 6290 génétique et dédevelopment研究所)对分子遗传数据库的帮助。
脚注
利益冲突:无声明。
- 收到2016年2月2日。
- 接受2016年8月9日。
- 版权©2016人队











{kind=link}
{kind=link}