





摘要
TGFBR2突变引起Loeys-迪茨综合征可呈现气胸http://ow.ly/SlMkS
到编辑:
原发性气胸影响人口的0.01%。10%的病例有气胸家族史,但在多数,一个明确的基因诊断不言。我们报告谁与左肺尖气胸提出了一个26岁,英国白人女性(图1一个).之前,她有偏头痛,右脚多处应力性骨折,近视,易瘀伤,腰椎侧弯,右侧髌骨自发性脱位。她以前没有气胸或任何其他呼吸系统疾病的病史,而且从未吸烟。
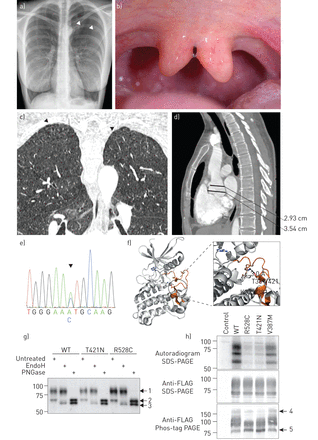
一)胸片示范左肺尖气胸。B)患者的悬雍垂的照片。C)胸部的冠状格式化计算机断层扫描(CT)(肺视窗设定)。箭头:在两个顶点众多胸膜下泡。d)胸主动脉的增强CT矢状重新格式化。黑色的lines shows maximal dimensions of the aortic root at the sinus of Valsalva (3.54 cm), confirmed by echocardiography, and sinotubular junction (2.93 cm). e) Chromatogram of sequence in the region of the c.1262C>A. Arrowhead: p.Thr421Asn mutation. f) Mutation site (enlarged in inset) modelled on crystal structure (PDB ID 2QLU) of activin receptor 2b (ActR2B). Residue numbering given in form ActR2B/transforming growth factor-β receptor II (TGFβRII). The mutation site occurs within the activation loop (orange) of the molecule that is critical for regulation of substrate binding and kinase activity. Thr361/421 forms a hydrogen bond (dashed line) with Lys323/381. Its perturbation would therefore destabilise the conformational behaviour of the activation loop. g) Protein glycosylation. Endoglycosidase (Endo)H and peptideN-糖苷酶(PNGase)酶切TGFβRII蛋白。人胚胎肾(HEK)293T细胞瞬时转染TGFβRII蛋白表达载体:野生型(WT)、新的T421N突变体和已知激酶死亡突变体R528C。蛋白裂解液或不处理,或用EndoH或PNGase消化。箭头1表示成熟糖基化,箭头2表示未成熟糖基化(内质网形式),箭头3表示完全去糖基化形式。注意,每种蛋白质中的大多数都对EndoH消化具有抗性,这表明高尔基体内的醣质处理。h)蛋白质自身磷酸化。将含有flag标记的TGFβRII蛋白(WT, T421N, R528C和V387M)的表达载体瞬时转染HEK293T细胞。细胞裂解,免疫沉淀法纯化FLAG蛋白。用FLAG多肽洗脱后,三分之一的材料与1µCi [γ3230˚C, 30 min, SDS-PAGE分离。剩余等量的洗脱液经SDS-PAGE或Phos-tag SDS-PAGE,转移到硝酸纤维素膜上,免疫印迹检测FLAG。箭头4表示缓慢迁移的磷酸化物种,而箭头5表示快速迁移的低磷酸化物种。
在检查中,她是过度活动(Beighton评分7/9),并有面肌,半透明的过度伸展的皮肤,在她的背部有条纹,胸壁不对称,双侧静脉曲张和扁平足。她的小舌裂(图1 b),患者上颚呈高拱形并有牙拥挤,臂宽/身高比增加(1.14)。在眼科诊所,点阵营养不良(周围视网膜虚弱易导致视网膜脱离)被确定为马凡氏综合征的眼部特征。患者胸部CT示根尖泡,超声心动图及CT示主动脉根部扩张(3.54 cm, Z-score >2) (图1 c和d).她59岁的母亲没有气胸,复查发现有轻度结缔组织疾病特征:皮肤伸展过度,关节活动过度(Beighton评分5/9),上颚高弓,轻度胸后凸,易瘀伤,复发性左肩脱位,裂孔疝,应力性失禁,左脚应力性骨折。
这些发现导致了Loeys-Dietz综合征(LDS)的临床诊断,这是一种影响转化生长因子(TGF)-β信号通路的常染色体显性疾病[1].LDS(类型I-V:在线人类孟德尔遗传号609192,610168,613795,分别614816和615582,)的特征在于血管发现(脑,胸,腹动脉瘤/夹层)和骨骼表现(漏斗胸或鸡胸,脊柱侧弯,关节松弛,arachnodactyly和马蹄内翻足)。大约受影响的个体的75%具有与LDS颅面表现(增宽,双歧悬雍垂/腭裂和颅缝早闭)型I;约25%具有LDSI的全身表现,但最小的或不存在颅面特征LDS II型。LDSI和LDSII形成临床连续统一体。The natural history of LDS is characterised by aggressive arterial aneurysms (mean age at death 26.1 years) and a high incidence of pregnancy-related complications, including death and uterine rupture. The diagnosis of LDS is based on characteristic clinical findings in the proband and family members and molecular genetic testing ofTGFBR1(TGF-β受体(TGFβR)I), TGFBR2(TGFβRII)、SMAD3 TGFB2(TGFβ2)TGFB3(TGFβ3)。
其他结缔组织疾病,如马凡氏综合征患者可能不会考虑,特别是面部粟丘疹和双歧悬雍垂的临床发现,因为这些是针对LDS。她TGFBR1和TGFBR2个基因从白细胞来源的DNA测序,她被发现是杂合的一个新的错义变体TGFBR2基因(c.1262C >;p.Thr421Asn;参考序列NM_003242.5) (图1 e).随后的测试发现她的母亲是这种变体的低水平嵌合体。
疾病相关基因的新错义变异的致病性通常是不确定的。421位苏氨酸是TGFβRII蛋白激酶结构域内高度保守的残基。在网上利用SIFT和Polyphen分析鉴定出的变异表明其具有致病性,SIFT预测这种变化会影响蛋白质功能,Polyphen预测这种变化是有害的。该变异既未在人类基因突变数据库中列出,也未在外显子组聚集联盟数据库中列出。
存在用于TGFβRII蛋白没有可用的晶体结构;然而,基于同源活化素受体的结构(ACTR)IIB(它们的激酶结构域内47.57%的同一性:TGFβRII残基244-544和残基的ActRIIB 190-480),我们评估了在这个高度保守区域的相互作用。该预测的新的突变会破坏分子内氢键稳定的高度保守的激酶活化环等影响活性(图1 f).这种方法不能排除对折叠的影响,尽管我们认为这不太可能。因此,我们评估了新变体的折叠和功能。人胚胎肾(HEK)293T细胞瞬时转染TGFβRII蛋白表达载体:野生型(WT)、新的T421N突变体和已知激酶死亡突变体R528C [2].蛋白质裂解物要么未经处理,要么被内质网蛋白质或肽上的未成熟糖苷酶H消化N-糖苷酶,它能清除所有N- 连接聚糖不论其复杂性。然后将裂解物进行SDS-PAGE,转移到硝酸纤维素和免疫印迹TGFβRII。所述变体T421N在培养的哺乳动物细胞中很好地表达和,类似于已知的无活性突变体R528C,显示了成熟的糖基化模式暗示它被很好折叠,并退出内质网(图1 g).
众所周知,TGFβRII突变体缺乏正常的激酶活性,但运输正常[2].因此,我们纯化了TGFβRII WT蛋白、T421N变体和激酶缺陷变体R528C。HEK293T细胞瞬时转染了编码TGFβRII蛋白的表达载体,在其c端标记FLAG: WT, T421N, R528C和一个弱形变体V387M(我们未发表的观察)。细胞裂解,免疫沉淀法纯化flag标记蛋白。用FLAG多肽从抗体中洗脱后,三分之一的材料与[γ]孵育32P]-ATP在30°C下处理30分钟,然后用SDS-PAGE分离,曝光于磷象片。正如预期的那样,阳性对照WT TGFβRII自磷酸化强烈,而阴性对照R528C没有图1 h).新颖的T421N突变体也显示自磷酸化受损,这表明它是致病性和确认LDS的临床诊断。由于放射性标记并不太适合于临床实验室实践中,我们也研究了PHOS标签胶未标记的蛋白质,然后进行免疫印迹。这种技术既分离通过它们的大小和磷酸化程度的蛋白质[3.].同样,阳性对照和阴性对照分离得很好,T421N突变体显示出明显的自磷酸化减少(下图)图1 h).
使单倍不足的精确机制TGFBR2导致疾病的原因尚不清楚,但在小鼠中,缺乏Tgfbr2在颅神经嵴细胞中导致TGFβ2和TGFβRIII表达升高[4].这增加了smad独立的下游信号,导致腭间充质增殖缺陷,可能是LDS中观察到的颅面缺陷的原因。
虽然有一项检讨指出,已知LDS的个案有气胸[5],据我们所知,这是第一个有记录的LDS合并气胸的病例。我们已经证实了致病突变是一种激酶死亡等位基因TGFBR2通过建立TGFβRII自身激酶活性的放射性和非放射性检测方法。随着通过靶向基因分析或通过大量基因的面板测试来识别多种变异成为常规,因此在体外用于推定致病突变的功能特征和验证的技术将变得越来越重要。LDS应该被认为是气胸的单基因原因,此外还有更广泛认识的原因:马凡氏综合征、血管性埃勒斯-丹洛斯综合征、结节性硬化/淋巴管平滑肌瘤病和Birt-Hogg-Dubé综合征[6].
我们的病人的气胸被保守治疗和解决。在随后的3年里没有再发生气胸。她开始使用氯沙坦和普萘洛尔作为主动脉保护,我们继续监测她的主动脉根部直径。总之,我们描述了一个以自发性气胸表现的LDS病例,它说明了在基因组时代功能验证的日益严峻的挑战,同时丰富了已知遗传疾病的表型谱,并证明了与相关疾病的重叠。
脚注
支持声明:S.J. Marciniak是英国医学研究理事会高级临床研究员。
利益冲突:没有宣布。
- 收到了2015年6月17日。
- 接受2015年8月24日。
- 版权©2015人队











![a) Chest radiograph demonstrating left apical pneumothorax. b) Photograph of patient's uvula. c) Coronal reformat of chest computed tomography (CT) (lung windows setting). Arrowheads: numerous subpleural blebs at both apices. d) Sagittal reformat of contrast-enhanced CT of thoracic aorta. Black lines shows maximal dimensions of the aortic root at the sinus of Valsalva (3.54 cm), confirmed by echocardiography, and sinotubular junction (2.93 cm). e) Chromatogram of sequence in the region of the c.1262C>A. Arrowhead: p.Thr421Asn mutation. f) Mutation site (enlarged in inset) modelled on crystal structure (PDB ID 2QLU) of activin receptor 2b (ActR2B). Residue numbering given in form ActR2B/transforming growth factor-β receptor II (TGFβRII). The mutation site occurs within the activation loop (orange) of the molecule that is critical for regulation of substrate binding and kinase activity. Thr361/421 forms a hydrogen bond (dashed line) with Lys323/381. Its perturbation would therefore destabilise the conformational behaviour of the activation loop. g) Protein glycosylation. Endoglycosidase (Endo)H and peptide N-glycosidase (PNGase) digestion of TGFβRII protein. Human Embryonic Kidney (HEK)293T cells were transiently transfected with expression vectors encoding TGFβRII proteins: wild type (WT), the new T421N mutant and a known kinase-dead mutant, R528C. Protein lysate was either left untreated or digested with EndoH or PNGase. Mature glycosylation is indicated by arrow 1, immature glycosylation (endoplasmic reticulum form) by arrow 2 and the fully deglycosylated form by arrow 3. Note the majority of each protein is resistant to EndoH digestion, indicating glycan processing within the Golgi apparatus. h) Protein autophosphorylation. HEK293T cells were transiently transfected with expression vectors encoding FLAG-tagged TGFβRII proteins: WT, T421N, R528C and V387M. Cells were lysed and FLAG proteins purified by immunoprecipitation. After elution with FLAG peptide, one third of the material was incubated with 1 µCi [γ32P]-ATP for 30 min at 30˚C then separated by SDS-PAGE. Equal amounts of the remaining eluate were subjected to SDS-PAGE or Phos-tag SDS-PAGE, transferred to nitrocellulose membrane and immunoblotted for FLAG. Arrow 4 identifies a slowly migrating, phosphorylated species, while arrow 5 indicates a rapidly migrating, poorly phosphorylated species.](http://www.qdcxjkg.com/content/erj/46/6/1832/F1.large.jpg?width=800&height=600&carousel=1){kind=link}
{kind=link}