





摘要
据报道,肺动脉高压(pH)的动物模型升高了物质P(SP)水平,并且已经显示SP的慢性施用来诱导pH值。在本研究中,分析了活性氧物质(ROS)作为SP诱导的血管重塑和pH的介质的作用。
通过用[SAR的治疗方法在精密切割肺切片中诱导血管重塑。9.,见面11.(o.2) - sp和缺氧。用功能分析用于研究慢性[SAR9.,见面11.(o.2)] - 上ROS,其参与PH的发病机理的增殖和产生SP介导的作用。非肽NK-1受体拮抗剂CP 96345被用于块[肌氨酸9.,见面11.(o.2) - sp效果。DichlorofloureesIndetate方法测定ROS产生和增殖,分别掺入5-溴-2'-脱氧尿苷。
1%的氧(5.8倍)或[Sar9.,见面11.(o.2) - 肺泡区中常氧中的sp(8倍)。[SAR.9.,见面11.(o.2) - SP没有进一步提高缺氧中的ROS水平,表明氧中依赖性机制。高ROS水平稳定缺氧诱导因子-1α并诱导小血管的增殖(4.3倍缺氧和[SAR9.,见面11.(o.2) - sp)。在CP 96345,NitroBluetazolium,CP 96345的存在下阻止了ROS产生和增殖。N-acetylcysteine-和diphenyleneiodonium。
本研究表明的结果表明SP在肺动脉高压血管重塑相关的增殖事件中的作用。
参与初级肺动脉高压(pH)和更常见的仲pH的发育病理学途径仍然不清楚。所有形式的pH的常见特征是肺血管重塑和右心室肥大。pH在人类pH的主要原因是肺泡缺氧1由于慢性肺部疾病导致继发pH值2。缺氧诱导包括肺血管平滑肌细胞(VSMC)在内的各种细胞中活性氧(ROS)的生成,并导致VSMC和外膜成纤维细胞的增殖3.。该机制涉及普遍表达的缺氧敏感转录因子缺氧诱导因子(HIF)-1α,其被ROS稳定1。HIF-1α激活在氧气上最大化(O.2)浓度为0.5-2%4.,而在常氧条件下,泛素促使途径迅速降解5.。
几种观察结果表明,物质p(sp)涉及病理生理事件,导致pH中观察到的血管重塑。稳定SP类似物的药理应用诱导血管重塑,导致介质增厚和大鼠pH的发育,同时已经显示出与TachykinnN-1受体拮抗剂CP 96345的预处理以防止SP诱导的pH值6.。在患有PH的患者中,SP不能引起在健康受试者中观察到的明显的内皮依赖性血管扩张7.。而血管扩张剂反应已被证明是通过激活NK‐1受体和释放NO和前列环素介导的8.,SP诱导的重塑机制仍不清楚。在肺血管的一官碱诱导炎症的大鼠模型中,建议ROS增加SP水平9.,可能通过灭活SP降解酶中性内肽酶(EC 3.4.24.11)。
由于缺氧和SP诱导的血管重塑和pH的相似性,在本研究中解决了SP通过产生ROS产生其效果的假设。使用小鼠的精密切割肺切片(PCLS)作为模型,研究了SP对ROS产生和HIF-1α稳定的影响,以及肺血管中增殖的影响。PLC将细胞培养物在有机体系中的优点结合在一起。它们主要用于药理学10.那11.或毒物学研究,并允许使用人体组织(回顾见12.)。
方法
组织处理
通过宫颈脱位杀死动物后,将500 u甲普丁(Hoffmannla Roche,Grenzachwyhlen,Green)和1毫米甘油林硝酸盐(Merk,Darmstadt,德国)注射到右心室中。然后将肺血管床灌注,用10ml含有0.2%丙基蒽氯化物(重量/体积; Merck),500 U肝素和10μM甘油硝酸盐的10ml预胶水床。然后用1%的低熔胶琼脂糖(德国慕尼黑BioRad)滴注肺部via将气管和胸内脏转移到冷却的林格中。使用vibraatome(Leica,Bensheim,德国)以250μm的厚度切割pcls。这些部分在最低基本培养基(MEM; Gibco,Heidelberg,德国)中培养,在38°C下没有额外的血清,含有21和1%的o2分别。在4小时后改变培养基以除去溶解的琼脂糖。
反应性氧物种测定
在两组使用二氯荧光素二乙酸酯的11只小鼠的pcl中测定了ROS的产生(DCF‐DA;Alexsis, Grünberg,德国),如前所述13.。在1μmDCF-DA存在下孵育六只动物的组1小时,用21或1%o孵育2。在两者o.2条件,5 nM [Sar9.,见面11.(o.2)] - SP,在非肽NK-1受体拮抗剂CP 96345(1μM)的存在下和不存在下,加入。Control sections were incubated with 1 µM CP 96345 or MEM alone. In a second set of experiments, the effects of ROS scavengers, 1 µM nitroblue tetrazolium (NBT; Sigma, Deisenhofen, Germany) and 1 µMN- 在常氧的条件下,仅在常氧的条件下在五只动物中检查了抑制主要ROS发电酶的五苯基碘(DPI;10μm; Calbiochem,Bad Sodene,Germany)的乙酰胞嘧啶抑制剂二苯基碘化物(DPI;10μm;
然后将这些部分固定在4%多聚甲醛(PFA)中,在4%磷酸盐缓冲盐水中洗涤20分钟,在4℃下在0.1M磷酸盐缓冲盐水中洗涤10分钟并保持在黑暗中。共聚焦激光扫描显微镜用于评估转化的DCF-DA的相对量,从而评估ROS的量。在薄壁的五个领域中,以致盲的方式量化每场的ROS产生细胞的数量。在相同水平的各个部分随机选择这些字段,但注意到没有看到大型气道或血管。
增殖试验
在含有1×10的血清红外MEM中孵育一组新的12只动物-4 M 5‐Bromo‐2′‐Deoxyuridine (BrdU) and gassed with 21 or 1% O272 h。在两者o.2条件,5 nM [Sar9.,见面11.(o.2) - 在存在和不存在1μmCC96345的情况下SP。另外,1μmnbt,1μmnac和10μmdpi的影响[sar9.,见面11.(o.2)] - SP在含氧量正常调节增殖活性进行了研究。
切片在4% PFA中固定45分钟,并包埋在TissueTek®(OCT化合物;Sakura Finetek Europe BV, Zoeterwoude, Netherlands),随后用低温恒温器(Leica, Weiterstadt, Germany) 10µm切片进行免疫组化。检测合并的BrdU需要双链脱氧核糖核酸的变性。切片在4°C 0.1 N HCl中孵育10分钟以去除组蛋白,在室温下转移到2n HCl中30分钟,用0.1 M硼砂(pH 8.5)中和10分钟。用单克隆抗体(DAKO, Hamburg, Germany)和小鼠免疫试剂盒(DAKO)观察BrdU合并,然后用strepavidin Texas Red (Amersham, Freiburg, Germany)观察BrdU合并,然后用多克隆兔vonWillebrand Factor抗体(DAKO)进行反染色,用异硫氰酸荧光素标记的山羊抗血清(Dianova, Germany)检测。Hamburg,德国)来识别脉管系统和4 ',6 -二氨基- 2 -苯吲哚,diactate (Sigma)来识别细胞核。在每个实验条件下,每只动物用荧光显微镜(DMRA2;徕卡,Weiterstadt,德国)在200倍放大。结果给出了每条血管的BrdU阳性核。
缺氧诱导因子1α的免疫组化定位
使用单克隆抗体(1:50; DPC Bieromann,Bad Nauheim,Germany)使用单克隆抗体(DPC Biermann,Bad Nauheim,Germany)来定位HIF-1α的平行抑制液部分。
统计分析
使用kruskalwallis分析数据,曼海夫尼U-test和p≤0.05被认为是显着的。错误被给出SEM。
结果
活性氧的生成
To determine the effect of SP on ROS generation, PCLS were incubated with DCF‐DA for 1 h under 21 and 1% O2后者作为ROS生成和血管重塑的后一种代替,作为缺氧下的ROS产量增加,并且慢性肺泡缺氧经常用于在动物模型中诱导pH值。五个nm [sar9.,见面11.(o.2) - 在存在和不存在1μmCCC96345的情况下,向培养基中添加1μmCCC96345。ROS生产在常见条件下非常低(图1⇓和2⇓)虽然缺氧导致肺泡区内的个体细胞中的ROS产生强大而显着增加(图1⇓和2⇓),但不是在较大的脉管系统,其使用相差显微镜鉴定。Sections incubated with SP displayed a similar ROS‐generation pattern under normoxic conditions as compared to hypoxia (figs. 1⇓和2⇓)但是,生产的细胞数量明显高于缺氧(图2⇓)。对SP和缺氧的主观细胞单独减少阳性细胞的数量(图2⇓)。in.cubating the sections with the NK‐1 receptor antagonist CP 96345 (1 µM) for 15 min prior to the administration of [Sar9.,见面11.(o.2) - SP显着减少了常氧基产生的ROS产生的细胞数量(图2⇓)但没有影响缺氧条件下的ROS生成(图2⇓)。单独对拮抗剂的应用对该系统中该系统中的ROS产生没有影响(图2⇓)。正如预测的那样,用NBT,NAC和DPI的肺切片的温育导致引起[肌氨酸DCF-DA或减少自由基的产生的完全抑制9.,见面11.(o.2)]‐SP在常氧条件下(图3a⇓)。
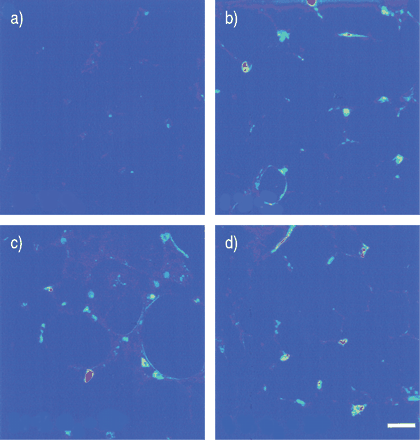
在21%氧(A和C),1%氧(B和D)和[SAR中的重要肺截面中的反应性氧物种的反应性氧物种(超过1小时)的图像9.,见面11.(o.2)]物质p(c和d)。秤杆=20μm。
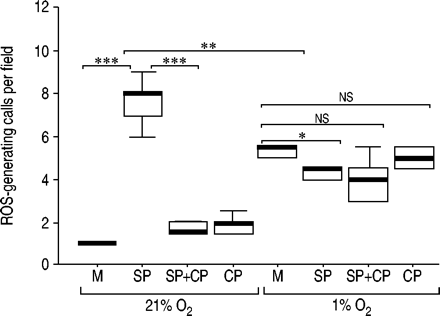
定量检测活性氧(ROS)生成细胞,平均每只动物5个电场(n=5)。M:中等;Cp: Cp 96345(1µm);SP:[SAR9.,见面11.(o.2)] - SP(5纳米)。***:P≤0.001;**:P≤0.01;*:P≤0.05;ns.:不重要的。
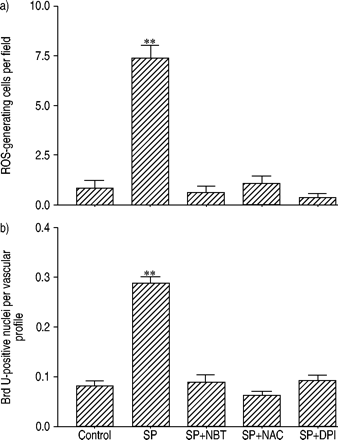
抑制物质P(SP)诱导的活性氧(ROS)产生(超过1小时)和增殖活性(以上超过72小时)通过自由基清除剂切割肺切片,降低烟酰胺腺嘌呤二核苷酸磷酸氧化酶抑制。a)每只动物的五个场的平均值定量ROS生成细胞(n = 5)。b)将增殖活性的定量为每实验条件和动物的25个田地(n = 5)。DPI:二苯基碘鎓(1μm);NBT:Nitroblue四唑(1μm);NAC:N- 缔酸盐(1μm);SP:[SAR9.,见面11.(o.2) - sp(5 nm)。**:p≤0.01。
增殖活动
切片在常氧条件下孵育导致小血管中低增殖活性(图4)⇓和5⇓),通过Brdu定量在72小时和随后的免疫组织中掺入核中。通过免疫组化检测vonfillebrand因素(数据未显示)鉴定小脉管系统。在同一段时间内使细分缺氧诱导小脉管系统(<50μm;图4)的巨大增殖增加⇓和5⇓),而较大的血管没有显示任何o下的任何增殖活动2浓度(>150μm;数据未显示)。五个nm [sar9.,见面11.(o.2)]‐SP在常氧条件下强烈诱导小血管介质内增殖(图4)⇓和5⇓),与缺氧时相同(图5)⇓)但是,[SAR9.,见面11.(o.2) - 在缺氧中施用时SP显着降低(图4⇓和5⇓)。在[萨9.,见面11.(o.2)] - 在常氧条件下的SP效果被强烈拮抗至几乎控制水平1μmCCC96345(图5⇓)。缺氧在[SAR造成的增殖降低9.,见面11.(o.2)] - 通过施用拮抗剂来逆转SP(图5⇓)。
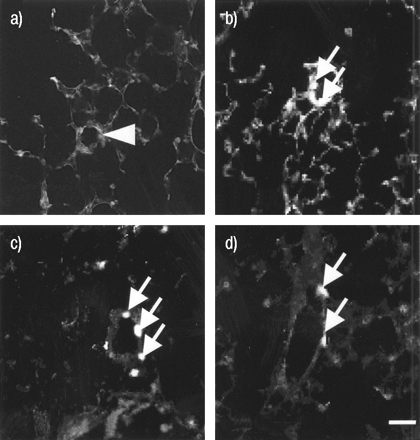
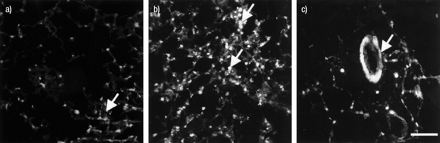
免疫组化定位5‐Bromo‐2’‐Deoxyuridine (BrdU)掺入检测肺小血管的增殖(超过72小时),在21%氧(a和c)、1%氧(b和d)和[Sar]的存在下9.,见面11.(o.2)]物质p(cand d)。箭头:Brdupositive核;箭头:船只。秤杆=20μm。
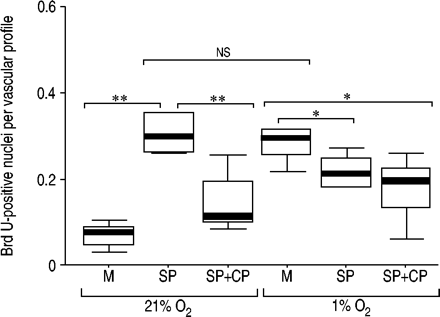
增殖的定量测定为平均每个实验条件和动物(N = 6)25个的字段。M:中等;Cp: Cp 96345(1µm);SP:[SAR9.,见面11.(o.2)] - SP(5纳米)。**:P≤0.01;*:P≤0.05;ns.:不重要的。
在[萨9.,见面11.(o.2) - 通过使用NBT或NAc清除ROS或通过DPI抑制黄曲霉素来完全阻断常氧条件下的SP诱导的增殖活性(图3B⇑)。
检测缺氧诱导因子-1α
正如预测的那样,在常氧条件下肺实质中无法检测到转录因子HIF‐1α(图6a)⇓)。Subjecting the sections to hypoxia stabilised HIF‐1α in cells of the lung parenchyma (fig. 6b⇓)。本地化仅限于核(图6B和C.⇓)。在肺部孵育[SAR9.,见面11.(o.2) - spunder normoxic conditions, HIF‐1α was detected in the parenchyma, as well as a small number of vessels (fig. 6c⇓),而较大的血管没有显示任何HIF-1α稳定(数据未显示)。
72小时后72小时后缺氧诱导因子-1α的免疫型诱导因子1α(21%氧气(o2))),b)缺氧(1%o2c)[sar9.,见面11.(o.2)]常氧中的物质p。箭头:船只。秤栏:50μm。
讨论
PH值的增殖事件是标志性病理特征。这些包括VSMC,成纤维细胞和内皮细胞的增殖,导致在pH中观察到的重塑14.。其结果是,在肺血管床异常增殖活性可以用作血管重塑的指示。
本研究已经解决了SP对扩散表明的ROS和血管重塑的产生的影响,而不试图在PCLS中表征增殖细胞,其显示在支气管中的睫状体击打纤毛,并且在72小时培养后药理学刺激(数据未显示)。这与M观察到的结果一致阿廷等等。10.。此外,已经表明,PCLS保持结构完整性长达60天,但内皮可能在7天后可能在其完整性下降15.。
SP对肺截面的应用导致ROS介导的血管重塑,其增殖活性增加表明。这与通过慢性治疗大鼠的结果获得的结果一致6.。如缺氧所观察到16.,PCLS的72小时培养中的SP治疗是研究肺动脉中血管重塑的有价值模型,这允许时间和动物数减少。
在一角质碱诱导的pH中,显示ROS促使SP的水平9.。该研究的结果表明,在PCLSTO严重缺氧或低剂量的曝光后产生的氧气基数是产生的[SAR9.,见面11.(o.2)] - 在(Peri)血管细胞中的1小时内。[sar的影响9.,见面11.(o.2) - 由于拮抗剂CP 96345显着减少了ROS发电单元的数量,因此SP被NK-1受体介导。使肺部肺截面缺氧和SP对ROS产生没有协同效应,表明SP引起的ROS生产是o2依赖。
Proliferation of vascular cells occurred only in small (<50 µm), resistancetype vessels, which is the part of the pulmonary vascular bed responsible for the pressure elevation observed in PH3.。缺氧导致增加的ROS产生在各种细胞中由任一膜结合还原的烟酰胺腺嘌呤二核苷酸磷酸(NADPH)氧化酶复合物17.那18.或增加线粒体ROS生成13.。已经显示NADPH氧化酶产生的ROS通过激活细胞外信号调节蛋白激酶(ERK)-2与C-FOS的后续表达,在人体主动脉的VSMC中诱导增殖viap21.RAS/ RAF-1 / MEK2路径19.,而线粒体强化RO通常导致细胞死亡20.那21.。事实上,缺氧NADPH氧化酶的激活已经VSMC描述22.。已显示低剂量的ROS在各种细胞中诱导增殖23.包括VSMC24.。
血管增生事件[Sar9.,见面11.(o.2)] - SP似乎也通过NK-1受体介导的,如CP 96345的给药做不仅抑制ROS的产生,而且血管的增殖。由于由[肌氨酸诱导增殖活性9.,见面11.(o.2- SP被活性氧清清剂NBT和NAC以及黄酮蛋白抑制剂DPI所阻断,似乎有可能对[Sar .]产生增殖反应9.,见面11.(o.2) - SP由ROS生成介导。
通过用NBT DPI orscavenging ROS membranerelated ROS产生的抑制已显示导致acomplete停止在缺氧诱导增殖thepulmonary脉管16.。此外,给予[SAR的大鼠的CP 96345是否抑制了大鼠pH的发育。9.,见面11.(o.2) - sp6.。相比于单独的缺氧,血管细胞的增殖是当下标[肌氨酸9.,见面11.(o.2) - 在缺氧中加入SP,表明稍微拮抗作用,这可能是Dueto激活不同信号转导途径的激活。ROS响应缺氧和[SAR的ROS引发的增殖事件9.,见面11.(o.2) - 根据常氧条件下的SP可能涉及HIF-1α的稳定性,因为通过免疫组织化学鉴定蛋白质。臭日中缺氧或[SAR中的妥善核中的血清中1α-阳性核的分布相同9.,见面11.(o.2) - SP孵育,但血管在与[SAR孵育的部分中展示了Strontoner免疫反应性。9.,见面11.(o.2) - SP,同样可能是由于不同信号转导途径的激活。
广泛表达的HIF-1α是缺氧基因表达的全局调节子虽然它被认为是与细胞typespecific调节因子相互作用,以唤起特定小区反应,如。促红细胞生成素或血管内皮生长因子的表达25.。ROS也可能导致诱导Protooncogenes CMYC和C-FOS26.,这反过来通过诱导细胞周期蛋白表达来激活细胞周期27.。其他研究表明,蛋白激酶C介导的磷脂酰肌醇3磷酸激酶/哺乳动物雷米霉素靶蛋白/p70的激活S6K.肺部VSMC中的激酶信号转导通路28.,这可以通过ROS被触发29.那30.。ROS也可能激活ERK‐5 (BMK‐1)via所述的c-Src激酶31.,它在缺氧时被激活,被认为是HIF‐1α的激活激酶32.。
总之,本研究提供了物质P对肺动脉高压病理学病理学涉及的重塑过程作用的依据。在此过程中,活性氧speciesinduced物质P水平的增加,如图中箭头C母鸡等等。9.和P物质介导的活性氧的产生,在本研究中所示,可能导致恶性循环延续肺动脉高压。将得到的血管重塑与阻力血管的受损血管扩张反应相关联。这些机制在本地进行,不涉及中央反射,在生活精密切割肺片观察疗效。
- 收到2003年3月13日。
- 接受2003年5月7日。
- ©ers Journals Ltd










![Pseudocoloured images of reactive oxygen species generation (over 1 h) in the parenchyma of vital lung sections in the presence of 21% oxygen (a and c), 1% oxygen (b and d) and [Sar9,Met11(O2)]substance P (c and d). Scale bar=20 µm.](http://www.qdcxjkg.com/content/erj/22/4/596/F1.large.jpg?width=800&height=600&carousel=1){kind=link}
{kind=link}
![Quantitative assay of reactive oxygen species (ROS)generating cells as a mean of five fields per animal (n=5). M: medium; CP: CP 96345 (1 µM); SP: [Sar9,Met11(O2)]‐SP (5 nM). ***: p≤0.001; **: p≤0.01; *: p≤0.05; ns: nonsignificant.](http://www.qdcxjkg.com/content/erj/22/4/596/F2.large.jpg?width=800&height=600&carousel=1){kind=link}
{kind=link}
![Inhibition of substance P (SP)‐induced reactive oxygen species (ROS) generation (over 1 h) and proliferative activity (over 72 h) in precision cut lung slices by radical scavengers and reduced nicotinamide adenine dinucleotide phosphate oxidase inhibition. a) Quantification of ROS generation cells as mean of five fields per animal (n=5). b) quantification of proliferative activity as a mean of 25 fields per experimental condition and animal (n=5). DPI: diphenylene iodonium (1 µM); NBT: nitroblue tetrazolium (1 µM); NAC: N‐acetycysteine (1 µM); SP: [Sar9,Met11(O2)]‐SP (5 nM). **: p≤0.01.](http://www.qdcxjkg.com/content/erj/22/4/596/F3.large.jpg?width=800&height=600&carousel=1){kind=link}
{kind=link}
![Detection of proliferation (over 72 h) in small pulmonary vessels assayed by 5‐Bromo‐2′‐Deoxyuridine (BrdU) incorporation localised by immunohistochemistry in the presence of 21% oxygen (aand c), 1% oxygen (b and d) and [Sar9,Met11(O2)]substance P (cand d). Arrows: BrdUpositive nuclei; arrow head: vessel. Scale bar=20 µm.](http://www.qdcxjkg.com/content/erj/22/4/596/F4.large.jpg?width=800&height=600&carousel=1){kind=link}
{kind=link}
![Quantitative assay of proliferation as a mean of 25 fields per experimental condition and animal (n=6). M: medium; CP: CP 96345 (1 µM); SP: [Sar9,Met11(O2)]‐SP (5 nM). **: p≤0.01; *: p≤0.05; ns: nonsignificant.](http://www.qdcxjkg.com/content/erj/22/4/596/F5.large.jpg?width=800&height=600&carousel=1){kind=link}
{kind=link}
![Immunohistological detection of hypoxia inducible factor‐1α after 72 h in sections subjected to a) normoxia (21% oxygen (O2)), b) hypoxia (1% O2) and c) [Sar9,Met11(O2)]substance P in normoxia. Arrows: vessels. Scale bar: 50 µm.](http://www.qdcxjkg.com/content/erj/22/4/596/F6.large.jpg?width=800&height=600&carousel=1){kind=link}
{kind=link}