





文摘
慢性血栓栓塞肺动脉高压(CTEPH)是一种罕见的、进步的肺血管疾病,通常是在急性肺栓塞的结果。CTEPH通常始于持续阻碍组织的大型或中型肺动脉血栓。血栓未能解决异常可能与纤维蛋白溶解或潜在的血液学的或自身免疫性疾病。现在也知道小血管异常造成血液动力学的妥协,在CTEPH功能障碍和疾病进展。小血管疾病可能发生阻塞的区域,可能引发的未解决的血栓性材料,和下游的遮挡,可能是因为过度的抵押品从高压支气管和系统性动脉血液供应。潜在的小血管疾病的分子过程是不完全理解和在这一领域还需要进一步的研究。小血管疾病的程度有一个实质性影响的严重性CTEPH和手术后的结果。介入和医疗CTEPH应该恢复正常的肺血管内的流量分布,卸载右心室和预防或治疗小血管疾病。它需要早期,可靠的识别患者CTEPH专家中心和使用最佳的治疗方法。
文摘
CTEPH复杂的病理生理学包括持续组织血栓和广泛的小血管疾病http://ow.ly/IgMH309hwmq
介绍
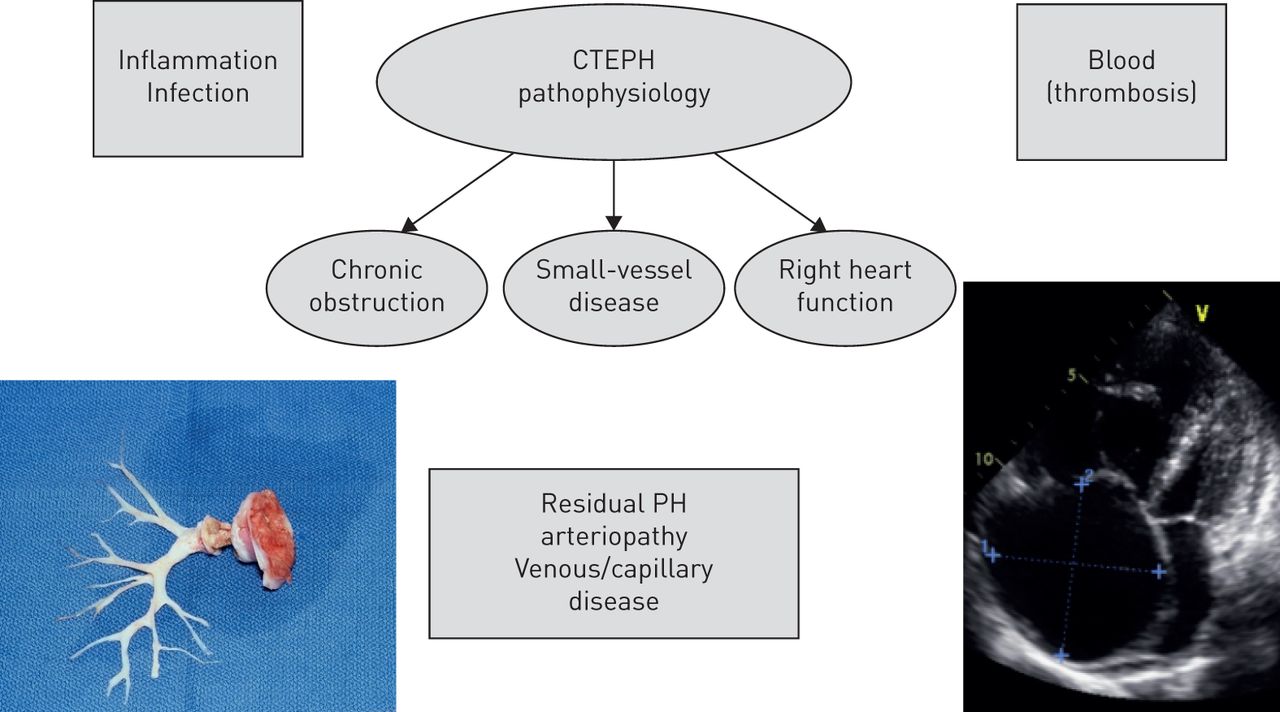
慢性血栓栓塞肺动脉高压(CTEPH)列为4组存在肺动脉高压的临床分类(1]。它是一种罕见的,进步的肺血管疾病有一个可怜的结果如果不及时治疗(2]。多年来它已经清楚CTEPH可能发生的并发症急性肺栓塞(PE)后静脉血栓栓塞(VTE) [3]。CTEPH肺动脉高压的机制是多因素疾病。最近的见解表明CTEPH涉及不仅持续组织在近端肺动脉血栓(主、大叶性和节段性),但也小血管疾病中扮演一个重要的角色在疾病的发展和进展(图1)[4]。在本文中,我们审查的最新进展对我们理解CTEPH的病理生理学。

病理生理学的慢性血栓栓塞肺动脉高压(CTEPH)。PH值:肺动脉高压。从[复制和修改4经允许)。
体育和CTEPH之间的关系
CTEPH通常被认为是一种罕见的和晚期并发症的一个或多个事件的急性PE没有解决尽管≥3个月的治疗抗凝。一大潜在国际CTEPH包括注册中心报道,75%的患者有急性PE (5]。然而,这个频率可能高估了,由于没有很好地诊断急性PE记录大量的情况下,可能条件以前记录的PE CTEPH的可能是第一个表现。事实上,不完整的决议的急性PE并不少见:一些研究报告,持续肺灌注缺陷在显像观察> 50%的情况下,3个月后的抗凝治疗(6]。幸运的是,大部分的这些患者不出现症状性肺动脉高压。
CTEPH可以开发几个月或几年之后急性PE(可能保持沉默),尽管继续抗凝,在缺乏新的症状或任何新的急性事件(7- - - - - -10]。
从德国和法国的肺动脉高压注册数据显示每年的发病率CTEPH每年四和超过一百万分之六的成年人,分别为(11友谊医院)(g . Simonneau服务de Pneumologie Bicetre,巴黎Sud大学,巴黎,法国;未发表的数据);这对应于∼300名新诊断的患者CTEPH每年在法国。的发病率CTEPH后急性PE尚未明确。前瞻性研究发表的诊断证实了右心catheterisation (RHC)的发病率CTEPH后症状急性PE据报道,从0.4%到6.2%不等(在线补充表S1) (7,10,12- - - - - -22),汇集发生率为3.4% (95% CI 2.1−4.4%)。考虑到∼30在法国每年诊断出000例急性PE (23),CTEPH发病率为3.4%每年会导致1000新CTEPH病例;远远超过实际上观察到的。大多数这些研究可能高估了CTEPH后急性PE的发病率。过高的原因之一是,很多患者已存在,未确诊的CTEPH PE的索引,当我们观察经常在日常实践。因此,在现实中,这些研究报告的发病率是事件和流行情况。在大多数研究中,CTEPH被诊断出几个月后指数体育,这是令人惊讶的,因为一般CTEPH发展数年的“蜜月期”后没有任何症状。只有一项研究中,由Guerinet al。(19),已妥善解决这一问题。146年急性PE患者与治疗抗凝治疗。在平均随访26个月,8的146例疑似CTEPH因为持续的呼吸困难和超声心动图发现异常,并证实了CTEPH RHC七个病人(4.8%,95%置信区间2.3−9.6%)。然而,在急性PE、索引的时间只有两个病人收缩期肺动脉压力(sPAP) < 50毫米汞柱。在剩下的五个病人,sPAP范围从62到102毫米汞柱;这种程度的急性PE sPAP不是兼容的第一,因为不适应右心室(RV)不能产生如此高的压力。因此可能CTEPH在场时的指数在这五个病人急性PE。证实了这个建议的审查他们最初经由电脑断层扫描,资深的专家,因为所有CTEPH确诊患者在随访中至少有两个初始条件的迹象表示。因此,累积发病率CTEPH急性PE后Guerinet al。研究并不是4.8%,但大多数∼1.5%(两个146)。这将给估计每年450新的CTEPH病例在法国,与每年300例类似的报道在法国注册表。鉴于CTEPH发病率低的急性PE后,系统的肺通气/灌注扫描检测的存在CTEPH后续的急性PE不推荐。
血栓在CTEPH nonresolution
在大多数PE患者,栓子发生的重要决议,随后恢复血流量和血流动力学参数(正常化24]。然而,在一小部分患者残余组织凝仍然附着在肺血管壁。为什么只有少数病人未能解决新鲜血栓和发展CTEPH急性PE后仍是一个谜。病理标本在急性PE和CTEPH是完全不同的:在急性PE、新鲜血栓是红色的,很容易脱离肺动脉壁主要由纤维蛋白网的红细胞和血小板。CTEPH,慢性血栓是黄色的、高度肺血管壁附着,并含有胶原蛋白,弹性蛋白,炎症细胞,re-canalisation血管,更很少,钙化(25]。组织和纤维化的残余血栓性物质(称为“乐队和网”在肺血管造影术)损害血液流动,并最终导致CTEPH的发展(图2)[2,24]。各种因素一直怀疑背后的失败解决血栓;下面讨论其中的一些因素。

自然历史的慢性血栓栓塞肺动脉高压(CTEPH)。体育:肺栓塞。从[复制和修改2经允许)。
临床条件诱发CTEPH
似乎使人容易罹患静脉血栓栓塞的一些特性差血栓CTEPH分辨率和后续发展。例如,似乎大肺栓塞的风险发展为CTEPH [2,24),也许是因为裂解系统缺乏处理能力达到和溶解血栓或阻止大栓子。其他功能似乎增加CTEPH进展包括复发性肺栓塞的风险和抗凝不足2,10]。然而,这些因素不能解释的发展CTEPH在大多数患者中,和其他机制必须参与。
的风险增加CTEPH已经与许多其他因素,如自身免疫性和血液学的障碍(26)和并发症,如多个并发症患者存在更频繁CTEPH比与肺动脉高血压(PAH) [27]。433年的一项研究比较CTEPH对254名患者与其他患者nonthromboembolic形式的肺动脉高压,ventriculo-atrial分流术和感染的起搏器,脾切除术,前静脉血栓栓塞(特别是复发性静脉血栓栓塞),non-O血型,出现红斑狼疮抗凝/ antiphospholipid抗体、甲状腺替代疗法和恶性肿瘤史都确认为携带的风险增加CTEPH [27]。
癌症
癌症患者血栓栓塞事件的风险增加,造成各种机制包括纤溶和凝血系统的激活,急性期反应,炎症和细胞因子的生产(28]。从欧洲数据库发现687名患者CTEPH (n = 433)和non-thromboembolic肺动脉高压(n = 254)支持一个恶性肿瘤史和CTEPH之间的联系(或3.76,95%可信区间1.47 - -10.43;(p = 0.005)27]。作者认为,足够强大的证据是,以保证调查CTEPH在历史的癌症患者肺动脉高压。
炎症和感染
似乎有炎症组件CTEPH发展水平较高的c反应蛋白(CRP)在患者与健康对照组相比,以及显著减少肺部动脉内膜切除术后CRP(豌豆)[29日]。然而,c反应蛋白升高不是特定于CTEPH, PAH患者水平也升高。最近的研究结果证实,c反应蛋白,以及白介素(IL) -10年,单核细胞趋化蛋白1,巨噬细胞炎症protein-1α和基质金属蛋白酶9 (MMP)明显升高患者CTEPH [30.]。此外,手术病人进行了样本豌豆含有大量的巨噬细胞,淋巴细胞和中性粒细胞,c反应蛋白之间的相关性和中性粒细胞聚集,MMP-9和巨噬细胞之间的积累。前瞻性分析八CTEPH患者的血清il - 6水平,引发,interferon-γ-induced蛋白质(IP) -10年,monokine interferon-γ和巨噬细胞炎性protein-1α是随着年龄的增长,sex-matched健康对照组相比显著升高(31日]。CTEPH患者,但不是那些特发性多环芳烃,IP-10水平(与成纤维细胞迁移和活化)与运动能力负相关,心输出量、心脏指数,而il - 6水平呈正相关,与肺血管阻力(PVR),右心房压力和水平N终端激素原的脑利钠肽。另一个炎症标记CTEPH追究一个连接是肿瘤坏死factor-α:水平升高患者CTEPH与控制相比,和豌豆后减少32]。
慢性感染的存在(如金黄色葡萄球菌CTEPH(患者)已被确认33),尽管它的相关性尚不清楚(34]。一项研究发现在六个thromboemboli收获葡萄球菌DNA豌豆从CTEPH ventriculo-atrial患者分流术(33]。作者表明,血栓感染触发CTEPH的发展。静脉血栓形成的小鼠模型,葡萄球菌感染延迟血栓决议与upregulation转变增长factor-β和结缔组织生长因子(33]。患者的回顾性研究CTEPH (n = 433)和non-thromboembolic肺动脉高压(n = 254),存在ventriculo-atrial分流或感染起搏器CTEPH发展是一个重要的危险因素(或76.40,95% CI 7.67 -10 351;p < 0.001) (27]。
为血栓nonresolution生物和遗传风险因子
一直怀疑患者血栓nonresolution有血凝过快因生物异常。有趣的是,经典的遗传性血栓形成的危险因素,如。蛋白质C, S和抗凝血酶不足,V和二世和突变因素,没有更频繁CTEPH患者比健康控制人口(35]。在这个前瞻性研究,只有antiphospholipid抗体的频率和狼疮抗凝CTEPH患者高于在特发性肺动脉高压患者。在前瞻性病例对照研究中,水平的提高凝血因子八世被确定与CTEPH 41%的病人,这是明显高于健康对照组和non-thromboembolic PAH患者(36]。在相同的研究中,水平的血管性血友病因子,一种糖蛋白的粘合剂,企稳和激活因子,显著增加患者与健康对照组相比CTEPH PAH患者,持续增加的患者经历了豌豆。
ADAMTS13 (disintegrin和金属蛋白酶与血小板反应蛋白1型图案,成员13),也称为血管性血友病factor-cleaving蛋白酶、血管性血友病因子大小的调节,在止血法起着根本性的作用。严重缺乏ADAMTS13引起血栓性血小板减少性紫癜(37]。在病例对照研究中,罕见的过剩和低频编码单核苷酸的变异ADAMTS13深静脉血栓形成患者被发现与匹配的控制;此外,这些患者表现出相对较低的等离子体水平的ADAMTS13活动(38]。
研究利用寡核苷酸微阵列比较肺动脉内皮细胞中基因表达CTEPH患者与正常对照组> 1600个基因中发现的不同调节或表达下调39]。调节基因包括引发,这与豌豆CTEPH[后血液动力学的不稳定性40]。其他患者的基因变异报道CTEPH包括血管紧张素转换酶基因多态性(41)和一个插入fibrinogen-α基因的多态性(42]。骨形成蛋白II型受体的突变(BMPR2)基因已经被报道在病人诊断为CTEPH [43]。然而,早些时候和更大规模的研究不支持的角色BMPR2突变CTEPH[的发病机制44,45]。其他的研究描述了组织因子基因表达增加(46),增加突变的频率与多环芳烃(47和差异表达小分子核糖核酸48,49]。
血型
CTEPH患者更常见血型A, B和AB。在一项研究中,77%的患者与CTEPH non-O血型相比有58%的患者多环芳烃(p = 0.003)36]。欧洲注册建议non-O血型显著预测诊断CTEPH(或2.09,95%可信区间1.12 - -3.94;(p = 0.019)27]。ABO血型的轨迹是静脉血栓栓塞的易感性位点和non-O运营商有静脉血栓栓塞的风险高于O运营商50]。
纤维蛋白原在CTEPH和纤溶异常
CTEPH似乎高患病率的患者血液中纤维蛋白原的分子异常(51),如纤维蛋白原Aα-Thr312Ala [52,53]。这种突变导致改性纤维蛋白凝块结构,包括增加交联α-chains [34]。其他杂合的多态性确定患者CTEPH包括β-chain突变P235L /γR375W, P235L /γY114H P235L,和α-chain突变L69H R554H [51]。最近,β15 42纤维蛋白原E链的片段被证明延迟血栓决议在活的有机体内(2]。每个纤维蛋白的共同特征异常患者到目前为止发现CTEPH是他们能够抵抗生理血栓溶解,从而影响血栓决议(2,54]。例如,一项研究比较从CTEPH患者和健康对照组发现,纤维蛋白原纤维蛋白从病人抗plasmin-mediated裂解与控制(55]。作者认为这是由于结构的改变影响可访问性纤维蛋白或纤维蛋白原血纤维蛋白溶酶乳沟网站。此外,有一个持久性的纤维蛋白结构主题(如。的N终点站β-chain)在肺血管内,作者推测可能参与发展从急性PE CTEPH (55]。在这项研究中,Olman等。(56)增加1型纤溶酶原激活物抑制剂和钝化反应的组织型纤溶酶原激活物观察,表明血浆纤溶系统在CTEPH完好无损。最近的一项研究调查的潜在作用thrombin-activatable纤维蛋白溶解抑制剂(TAFI),肝脏产生的等离子体羧肽酶抑制剂,抑制纤维蛋白溶解的病理学CTEPH [57]。血浆TAFI水平和释放TAFI CTEPH患者血小板明显高于PAH患者或控制。此外,TAFI水平显著相关,抗血栓溶解在遗传分析和他们气球肺血管成形术后保持不变。这些观察表明一个重要的角色在病理生理学TAFI CTEPH。
血小板功能在CTEPH
观察platelet-activating条件如甲状腺激素替代疗法和脾切除术是风险因素CTEPH表明血小板的作用在其《创世纪》(5,27]。受损的小鼠模型研究血栓分辨率显示脾切除术后的初始体积增加血栓是由于血小板激活(58]。同一项研究报道在splenectomised血小板微粒的增加与non-splenectomised CTEPH病人。与控制相比,CTEPH患者血小板计数减少,高的平均血小板体积,增加自发性血小板聚集,降低血小板聚集反应受体激动剂(59]。这些观察表明凝血状态CTEPH患者血小板高营业额。Yaoita等。(60)报道,从CTEPH或PAH患者血小板被激活与肺动脉以外高血压控制P-selectin上衡量表面表达时,起始位点绑定和GTP-bound GTPase RhoA,参与血小板聚集。患者手术材料提取的豌豆CTEPH包含水平的提高血小板因子4,由血小板释放在受伤的网站61年]。这些观察表明CTEPH血小板功能障碍的病理作用。
受损的血管生成
研究在动物模型的受损血栓决议指出,受损的血管生成和recanalisation血栓可能参与的病理生理学CTEPH [58,62年]。受体蛋白激酶的内皮特异性删除插入域(flk-1)切除血栓血管生成和延迟血栓决议在人类深静脉血栓形成的小鼠模型62年]。缺乏血管豌豆标本建议缺乏血管闭塞血管生成是一个关键机制改造后静脉血栓栓塞(62年]。这些发现表明,医疗条件与CTEPH有关,如脾切除术、感染或异常磷脂物种,可能损害早期血栓血管生成,血栓决议中迈出的重要一步2,62年]。此外,水平的提高angiostatic因素,如血小板因子4,胶原蛋白I型和IP-10已确定从CTEPH患者和外科豌豆材料与血管生成减少和/或扩散和迁移,recanalisation不足可能导致血栓性材料(61年]。
血小板内皮细胞粘附molecule-1和血栓形成
血小板内皮细胞粘附分子(PECAM) 1是一个糖肽受体表达在血小板,内皮细胞和其他细胞类型。参与白细胞轮回,对炎症的刺激的反应,静脉血栓的关键组件决议(63年]。在一个小鼠模型模仿人类的深静脉血栓形成,PECAM-1不足导致显著更大的血栓和误导血栓决议(64年]。此外,人类尚未解决的深静脉血栓形成标本显示PECAM-1裂解形成的积累,和延迟血栓患者解决等离子体水平有了显著提高可溶性裂解PECAM-1相比那些血栓解决(64年]。白色和红色血栓患者从CTEPH显示减少PECAM-1表达式与unthrombosed血管相比,暗示PECAM-1不足的病理CTEPH [62年]。
小血管疾病CTEPH
组织学和机械方面
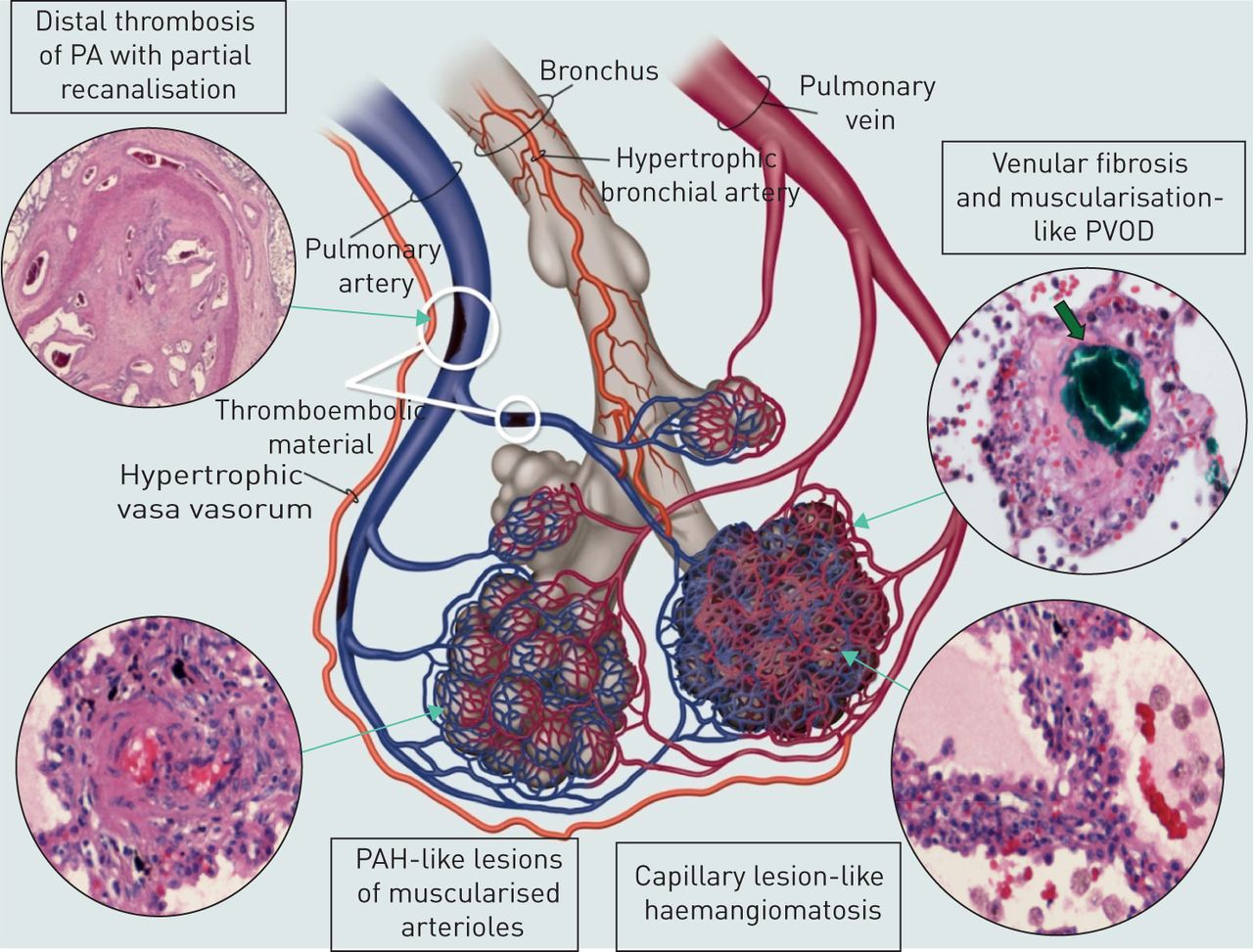
的闭塞近端(主、大叶性和节段性)肺动脉组织纤维化的血块是初始引发CTEPH发展。然而,它并不是唯一的肺动脉高压的机制在此设置。越来越多的证据表明,除了机械梗阻近端动脉,或多或少有些病人发展严重肺microvasculopathy(小血管疾病),首先描述了M奥泽和Bloor(65年在获得的肺组织活检或尸检。病理研究M奥泽和Bloor和其他作者披露全面特发性肺动脉高血压病变所观察到的类似的多环芳烃,其中包括内膜的增厚和肺阻力血管的重塑,古怪的内层的纤维化,内层的纤维肌性的扩散和丛状的病灶2,65年,66年]。这个血管重塑的影响远端肌肉的墙肺动脉(0.1−0.5毫米直径),甚至可能达到小动脉和小静脉的直径小于0.1毫米。这些变化是肺的经典解释为再分配流入nonoccluded肺动脉暴露于高压力和剪切应力,导致内皮功能障碍,逐步增加PVR和最终CTEPH症状。然而,这microvasculopathy不仅观察肺地区由nonoccluded肺动脉近端,而且远侧地肺动脉锢囚被纤维材料。这使它不太可能再分配的流量仅在肺动脉床能解释这种改造。Dorfmuller等。(67年)发现大型体循环和肺动脉循环(之间的吻合通过肥厚性支气管动脉和血管滋养管)CTEPH患者。有人推测,预先存在的吻合是打开支气管动脉之间的压力梯度,postobstruction肺动脉。这种机制可以帮助维持灌注和支持缺血性组织下游近端阻塞,但肺动脉循环的接触高压体循环可能导致一些病人肺动脉血管重塑,尤其是慢性血栓栓塞梗阻远端。在上述研究中,Dorfmuller等。观察到的重要反应,nonthrombotic微血管阻塞地区改造。墨水喷射实验在人类身上CTEPH和实验CTEPH显示小血管疾病的小猪模型不局限于前毛细管的小动脉,但另外担心postcapillary小静脉和小静脉。事实上,吻合已知存在体循环和肺静脉和肺毛细血管,可能导致病变,可能是类似于毛细管haemangiomatosis和肺静脉阻塞疾病(图3)[67年]。工作还需要解释完全是小血管疾病的发展和进步。

Microvasculopathy在慢性血栓栓塞肺动脉高压肺小动脉、小静脉和毛细血管。吻合的示意图表示系统性和肺循环之间通过肥厚性支气管动脉和血管滋养管。PA:肺动脉;PVOD:肺部静脉阻塞疾病;多环芳烃:肺动脉高血压。从[复制和修改67年经允许)。
小血管疾病CTEPH也包括远端血栓形成,在极少数情况下。病变可以扩散,可能当小肺小动脉远端近端完成障碍物不保持开放因为支气管动脉,吻合失败来培养。除了典型的CTEPH发现,这些患者的肺血管造影显示扩散的一个特殊方面,可怜的胸膜下毛细血管灌注的阶段(p . Dartevelle法国国家参考中心肺动脉高压,友谊医院Bicetre,比塞特尔勒,法国;个人通信)(图4)。

不实用的慢性血栓栓塞肺动脉高压患者显示一个典型的方面的胸膜下毛细血管阶段的低灌注肺血管造影术。图片请提供的p . Dartevelle法国国家参考中心肺动脉高压,友谊医院Bicetre,法国-比塞特尔勒。
小血管疾病的分子机制
最近的证据表明一氧化氮(NO)的参与可溶性鸟苷酸环化酶(国网公司)环鸟苷酸(cGMP)通路CTEPH的病理生理学。没有由血管内皮抑制血小板聚集和平滑肌细胞的增长68年,69年]。还没有激活国网公司合成cGMP,第二信使和许多行为包括平滑肌松弛。等离子体的不对称dimethylarginine水平,没有合酶的抑制剂,在CTEPH患者增加与控制(70年,减少内源性PAH患者没有发现水平和CTEPH71年]。CTEPH可能表明功能障碍的血管重塑抗增殖机制,包括没有途径。临床和血液动力学的改进与国网公司刺激患者riociguat CTEPH资格豌豆或持续性肺动脉高压术后复发还建议NO-sGC-cGMP通路的一个重要的角色的病理条件(72年- - - - - -74年]。
内皮素(ET) 1水平升高在CTEPH患者和动物模型的条件75年- - - - - -79年]。最近的证据显示一个潜在的角色ET-1慢性血栓内的平滑肌细胞增殖CTEPH和小血管疾病(80年]。血管内皮生长因子水平明显降低患者在豌豆CTEPH [81年]。水平的提高而,信号分子参与血管生成和平滑肌细胞增殖,已确定患者的肺部CTEPH [82年)和高术前angiopoietin-2水平与较差的预后相关的豌豆(83年]。初步研究CTEPH在小猪的典范84年)表明,小血管疾病与ET-1和il - 6基因表达的变化(85年]。
小血管疾病的临床意义
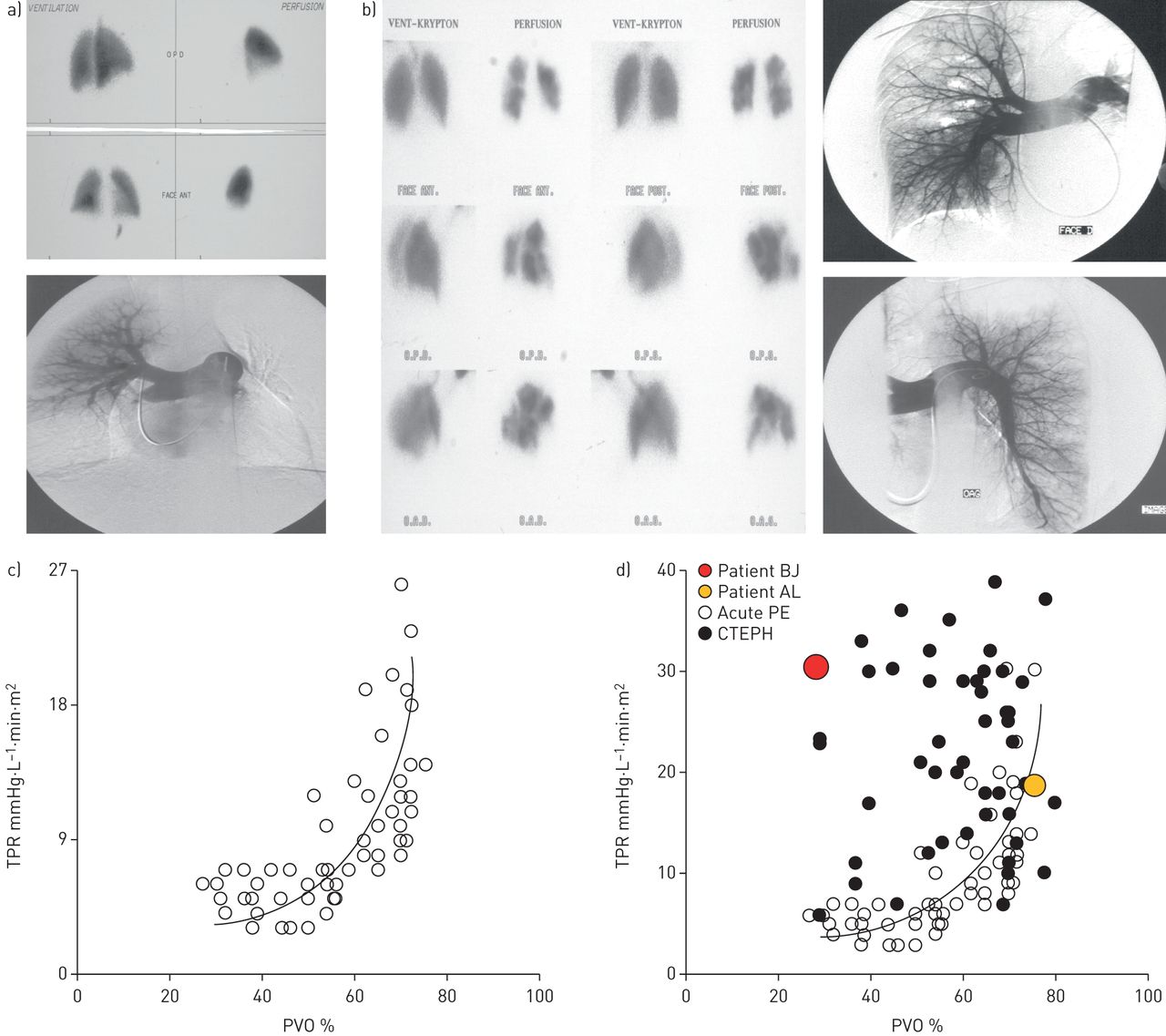
这些微血管的逐步发展变化可以解释为什么有些患者CTEPH恶化即使没有复发性肺栓塞。这种小血管疾病的严重程度是患者个体之间高度可变。广泛的存在小血管疾病患者应怀疑当机械阻塞的程度由近端纤维凝块(如。适度阻碍基于评估使用肺灌注扫描和/或肺动脉血管造影)不与PVR的大幅增加(86年(例子所示图5)。这些患者术后死亡率的风险更高87年,88年],小血管疾病不适合豌豆豌豆后有助于持续肺动脉高压,也不完全切除手术后近端血栓性材料(89年- - - - - -92年]。因此,对于优化疾病管理至关重要的检测和治疗CTEPH小血管重塑发生之前。广泛的小血管疾病对病人预后的不利影响手术后患者的前瞻性研究证实了26 CTEPH [91年]。上游电阻被分析评估手术肺阻塞RHC波形。上游阻力显著相关,术后肺总阻力指数和平均肺动脉压(PAP),和所有四个术后死亡患者在研究术前上游电阻< 60%。

肺血管阻力之间的关系(PVO)和肺血管阻力在慢性血栓栓塞肺动脉高压(CTEPH)和急性肺栓塞(PE)。与CTEPH)病人艾尔:24岁的女性。PVO的比例估计在肺灌注扫描为75%。左肺梗塞和闭塞的中下游叶。血液动力学:平均肺动脉压(PAP) 32毫米汞柱;心脏指数1.7 L·分钟1·米2;总肺血管阻力(TPR) 18.8毫米汞柱·L1·敏·米2。尽管75% PVO, TPR只有18.8。与CTEPH b)病人BJ: 54岁的女性。PVO的比例大约在35%,肺灌注扫描多个双边节段和subsegmental灌注缺陷。血液动力学:意味着PAP 45毫米汞柱;心脏指数1.4 L·分钟1·米2;TPR 32.1 mmHg·L1·敏·米2。c)的比例关系PVO评估急性PE患者肺灌注扫描和TPR (n = 31)。强大的双曲相关性被发现。d)对于一个给定的PVO的程度,大多数患者CTEPH (n = 45) TPR值高于急性PE患者(n = 31),这表明,除了机械由组织凝块阻塞小血管疾病。病人半岛位于双曲相关性(没有microvasculopathy),而病人BJ不成比例和高水平的TPR轻度PVO相比(严重microvasculopathy)。从[复制和修改86年经允许)。
发现小血管疾病引起的远端血栓形成(如前所述)也往往是严重的和与豌豆后的不良预后显著相关93年]。有趣的是,存在扩张的支气管动脉和肺动脉内吻合术循环,经常出现在CTEPH和防止远端血栓形成,与良好的生存和血液动力学的主要改进后豌豆(94年]。
以及不适合豌豆,小血管疾病患者无法接近CTEPH气球肺血管成形术,这或许可以解释这个介入过程后的持续肺动脉高压患者。组织纤维化的凝块狭窄远端肺动脉可以使用气球气球肺血管成形术治疗1 - 10毫米直径。
房车在CTEPH功能障碍和衰竭
CTEPH的特点是慢性房车后负荷增加和墙的压力。最初,这些负担导致房车房车肥大的特点是增加壁厚和细胞大小通过增加观察的(95年,96年]。这个过程的结果心肌细胞的内在能力感知和响应机械负载(96年]。起初,房车壁厚的增加导致墙压力和泵效率的提高降低了卸载个人的肌肉纤维(96年),和房车功能更像左心室(97年]。这些变化被称为“自适应”改造。这个过程不仅限于CTEPH患者,患者也出现在人民行动党< 25 mmHg和慢性血栓栓塞疾病(合法)97年]。这是演示了使用电导导管在一项研究中,患者之间的差异缝合法,CTEPH压力-容积循环形态学观察和控制,特别是在收缩期喷射(97年]。患者的海星,这些变化导致开发专属的房车后负荷升高血液动力学的肺动脉高压的定义。此外,RV放松更慢患者的海星和CTEPH与控制。作者认为这一特点房车慢性升高的后负荷,降低动脉合规。特发性肺动脉高压患者比较,近端CTEPH (pre -和参与)和远端(瘫痪)CTEPH,那些CTEPH明显短时间常数的肺循环比多环芳烃(98年]。这是符合建议从一个小研究,患者CTEPH房车收缩性和部分区域变化低于特发性多环芳烃或Eisenmenger综合症患者(虽然CTEPH患者(平均年龄50.8岁)以上比较器组(平均年龄42.2岁和41.2岁特发性多环芳烃和Einsenmenger综合征))(99年]。
前面描述的适应性变化可以维护房车功能在一段时间内,但RV并不能够维持长期超负荷的压力,从而增加身体活动期间进一步。此外,逐步改造的最初专利肺小动脉的床对RV不断增加负担,导致其不适应的改造。这是特点是古怪的肥大,房车扩张,房车收缩力降低,舒张功能不全和心肌纤维化96年]。房车扩张壁张力增加,需氧量增加,并减少灌注,导致恶性循环进一步妥协的收缩和扩张。房车中风减少体积,降低左心室肺动脉流和填充不足导致全身性低血压和房车冠状动脉灌注的恶化。进步的障碍,最终导致房车失败,CTEPH[死亡的主要原因95年,96年]。房车失败的时间进程,以应对病人之间的压力过载变化很大,可能由于压力的变化,负载、表型和神经体液的超速档(95年,96年]。虽然发展房车失败是明显要慢得多,类似的事件序列发生在急性PE在许多方面。最重要的两个差异是进步房车后负荷增加引起肺小动脉的正在进行的改造和适应房车心肌肥大的可能性,在急性PE缺席。是否增加了房车的质量是一个独立的危险因素的后续发展进步的房车失败CTEPH尚不清楚(96年]。
房车舒张功能不全,包括增加刚度、填充受损和长时间isovolumic放松,弥漫性心肌纤维化和sarcomeric加劲,相对可能发生肺动脉高压患者在疾病的早期可以表现出房车舒张功能受损而房车收缩功能相对保存(96年,One hundred.,101年]。
在肺动脉高压的动物模型、干扰血管生成和毛细管膨胀发生在功能失调的房车肥厚性组织,和房车心肌代谢开关从mitochondria-based脂肪酸氧化糖酵解(96年]。与控制相比,肺动脉高压患者表现出降低房车收缩运动和肺血管扩张性的损失(96年,102年]。
肺动脉高压的动物模型表示显著增加在不适应的房车肥大组织纤维化,相对于水平在适应性肥大96年,103年,104年]。纤维化在不适应的肥大是upregulation产生的病理过程和生长因子之间的相互作用(包括转换增长factor-β和结缔组织生长因子),激素和基质金属蛋白酶(96年]。
豌豆CTEPH患者改善房车功能,表明可以扭转病态改造(105年- - - - - -108年]。功能改进需要时间来发展,与血液动力学的变化,并立即(105年- - - - - -108年]。收缩压的降低房车墙后应力豌豆是一个关键因素在实现同步模式的左心室和房车峰值压力(106年]。气球肺血管成形术也可能改善房车功能(109年- - - - - -111年建立了),但不如豌豆(112年]。
房车的状态的患者经历了成功的豌豆或气球肺血管成形术治疗CTEPH尚不清楚。应该这样一个卸载房车被视为“先决条件”,因此会更好的维持一个潜在的后负荷增加,或者,相反,是不可逆转的损坏和不正常吗?有一个“临界点”RV在CTEPH引起的一系列事件吗?澄清这些问题可能会影响我们的策略和时间的治疗干预措施。
摘要和结论
CTEPH当前的理解已经超越了一种慢性梗阻引起的未解决的血栓性材料和合成房车功能障碍包括一个复杂的疾病包括慢性梗阻近端被纤维凝块,小血管疾病和改造整个肺血管床(4]。剩余血栓形成的过程坚持PE患者,这种残余血栓导致CTEPH,并不完全理解,但炎症和感染被认为扮演一个角色。小血管疾病的程度有一个实质性影响的严重性CTEPH和手术后的结果,因此小血管疾病的评估可以帮助确保最优管理病人。潜在的小血管疾病的分子进程并不完全清楚,但迄今为止见解给了线索的潜在机制的好处医学疗法。进一步研究小血管疾病患者CTEPH可以帮助识别那些将受益最多的三个管理策略目前气球(豌豆、药物治疗和肺血管成形术)并导致的发现新的治疗方法防止发展为不可逆转的房车失败。澄清这些问题可以改善我们的管理策略和治疗干预的时机。
补充材料
补充材料
请注意:补充材料并不是由编辑部,编辑和上传已由作者提供。
表S1。发病率CTEPH经右心catheterisation后急性PE呃- 0112 - 2016 - _supplementary_information
披露的信息
确认
编辑提供的援助是艾德菲通讯有限公司(英国Bollington),由拜耳公司(德国柏林)。
脚注
可以从本文的补充材料err.ersjournals.com
利益冲突:披露可以找到与这篇文章err.ersjournals.com
出处:出版同行评议的这篇文章是由拜耳公司赞助,柏林,德国(主要赞助商,欧洲呼吸审查问题143)。
- 收到了2016年12月2日。
- 接受2017年2月17日。
- 版权©2017人队。
犯错的文章都是开放和分布式根据创作188滚球软件共用署名非商业性4.0许可证。











![Pathophysiology of chronic thromboembolic pulmonary hypertension (CTEPH). PH: pulmonary hypertension. Reproduced and modified from [4] with permission.](https://err.ersjournals.com/content/errev/26/143/160112/F1.large.jpg?width=800&height=600&carousel=1){kind=link}
{kind=link}
![Natural history of chronic thromboembolic pulmonary hypertension (CTEPH). PE: pulmonary embolism. Reproduced and modified from [2] with permission.](https://err.ersjournals.com/content/errev/26/143/160112/F2.large.jpg?width=800&height=600&carousel=1){kind=link}
{kind=link}
![Microvasculopathy in chronic thromboembolic pulmonary hypertension involving pulmonary arterioles, venules and capillaries. Schematic representation of anastomosis between systemic and pulmonary circulation through hypertrophic bronchial arteries and vasa vasorum. PA: pulmonary artery; PVOD: pulmonary veno-occlusive disease; PAH: pulmonary arterial hypertension. Reproduced and modified from [67] with permission.](https://err.ersjournals.com/content/errev/26/143/160112/F3.large.jpg?width=800&height=600&carousel=1){kind=link}
{kind=link}
{kind=link}
{kind=link}
![Relationship between pulmonary vascular obstruction (PVO) and pulmonary vascular resistance in chronic thromboembolic pulmonary hypertension (CTEPH) and acute pulmonary embolism (PE). a) Patient AL: 24-year-old female with CTEPH. Percentage of PVO estimated on perfusion lung scan at 75%. Total occlusion of left lung and occlusion of right middle and lower lobes. Haemodynamics: mean pulmonary arterial pressure (PAP) 32 mmHg; cardiac index 1.7 L·min-1·m-2; total pulmonary vascular resistance (TPR) 18.8 mmHg·L-1·min·m2. Despite 75% PVO, the TPR was only 18.8. b) Patient BJ: 54-year-old female with CTEPH. Percentage of PVO estimated on perfusion lung scan at 35%; multiple bilateral segmental and subsegmental perfusion defects. Haemodynamics: mean PAP 45 mmHg; cardiac index 1.4 L·min-1·m-2; TPR 32.1 mmHg·L-1·min·m2. c) Relationship between percentage of PVO assessed by perfusion lung scan and TPR in patients with acute PE (n=31). A strong hyperbolic correlation was found. d) For a given degree of PVO, most patients with CTEPH (n=45) have higher TPR values than patients with acute PE (n=31), suggesting that, in addition to mechanical obstruction by organised clots, they have small-vessel disease. Patient AL is located on the hyperbolic correlation (no microvasculopathy), whereas patient BJ has a disproportionate and very high level of TPR compared to mild PVO (severe microvasculopathy). Reproduced and modified from [86] with permission.](https://err.ersjournals.com/content/errev/26/143/160112/F5.large.jpg?width=800&height=600&carousel=1){kind=link}
{kind=link}