





摘要
慢性血栓栓塞性肺动脉高压(CTEPH)是一种危及生命的疾病,由持续有组织的血栓阻塞肺动脉引起,与一次或多次急性肺栓塞发作有关。迄今为止,CTEPH的发病机制仍未得到解释。肺内膜切除术可以清除肺血管的阻塞,可以治愈患者。然而,一些不可及的远端肺阻塞和/或相关的远端肺血管病变可引起持续性肺动脉高压,这是主要的术后并发症。
CTEPH的病理生理学尚不完全清楚,提高对这种疾病的认识可以改善我们未来的手术和医疗管理。几十年来进行的许多尝试都未能在动物身上重现这种慢性疾病。然而,一些动物模型为CTEPH的病理生理和发病机制提供了新的见解。在这里,我们回顾了所有的动物模型,提高了对CTEPH的理解,并为进一步的研究提供了希望。这篇简短的综述分析了所有可用来研究CTEPH病理生理学的动物模型的优缺点。
关于Dana Point 2009年临床分类[1],慢性血栓栓塞性肺动脉高压(chronic thromboembolic pulmonary hypertension, CTEPH)是肺动脉高压(PH)的一种4型亚型,其中肺内膜切除术(pulmonary endarterectomy, PEA) [2可有效预防右心室衰竭导致的死亡。CTEPH是由于一次或多次急性肺栓塞期间形成的持续有组织的血栓阻塞肺动脉所致。血栓持续的原因尚不清楚,CTEPH的发病机制仍未解释。迄今为止,由于缺乏预测急性肺栓塞演变为CTEPH的危险因素,不允许制定预防护理和/或筛查计划。
肺血管阻力的增加被认为是近端肺动脉阻塞和远端肺血管病变共同作用的结果[3.,4],这些数据必须在PEA前进行量化,以评估风险并预测手术成功。然而,梗阻性和非梗阻性周围血管床病变发展的机制仍然未知。因此,由于缺乏有效的治疗方法,无法手术的CTEPH或可手术的伴有重要远端血管病变的CTEPH的医疗管理仍然很麻烦。
为了阐明CTEPH的病理生理学,在试图开发可靠的动物模型方面已经花费了相当大的努力。急性肺栓塞很容易在几种动物中发生。相比之下,诱导一种复制人类CTEPH所有成分的疾病已被证明具有挑战性。这些因素包括凝块的持久性和组织,肺动脉高压,由未解决的腔内物质引起的慢性肺动脉阻塞,缺血性肺区域的全体性血液供应的发展,通畅区域的肺血管病变,以及右心室重塑。在此,我们回顾了国际文献中描述的用于研究CTEPH发病机制和病理生理学的所有动物模型(表1).
持续性血管内血栓形成的动物模型
肺栓子或血栓不能溶解,而是组织成闭塞性纤维化物质的机制尚不清楚。假设包括诱发肺内皮细胞异常[42]和血管修复过程中的损伤[43]。一个角色原位与内皮细胞功能障碍相关的血栓形成可能解释了为什么高达63%的CTEPH患者没有记录在案的急性肺栓塞史[3.]。这一假设尚未在动物中进行研究,因为迄今为止尚未在患有CTEPH的人类中发现内皮细胞异常。相比之下,血栓组织过程已在低流量诱导下腔静脉血栓形成的可靠动物模型中进行了研究(表2).这些动物模型大多是在啮齿动物中开发的,目的是研究静脉血栓的消退[5- - - - - -22]。静脉瘀血可单独诱发血栓形成,也可合并诱发的高凝或机械性内皮损伤。K盎et al。[13]研究了颈静脉狭窄和机械性内皮损伤后的仔猪颈静脉血栓形成模型[15]。下腔静脉闭塞也曾在猴子身上进行过研究[15]。然而,小鼠和大鼠仍是使用最广泛的动物,因为它们具有成本效益和效率[7]。低流量啮齿动物模型的特征是层状血栓在3-4周内消退通过需要炎症细胞募集和血管生成信号的再通过程[5- - - - - -11,17- - - - - -22]。
在临床危险因素的研究中,CTEPH既不与与静脉血栓栓塞相关的经典血浆因子异常相关,也不与纤维蛋白溶解障碍相关。然而,有心室心房分流或心脏起搏器感染史会增加CTEPH的风险[44,45]。这一发现促使了对动物模型的研究。因此,Bondermanet al。[23]利用下腔静脉狭窄和内皮损伤引起的小鼠低流量静脉血栓形成模型,研究葡萄球菌感染在延迟血栓消退和促进纤维化前分子表达中的作用。
双血管腔室理论
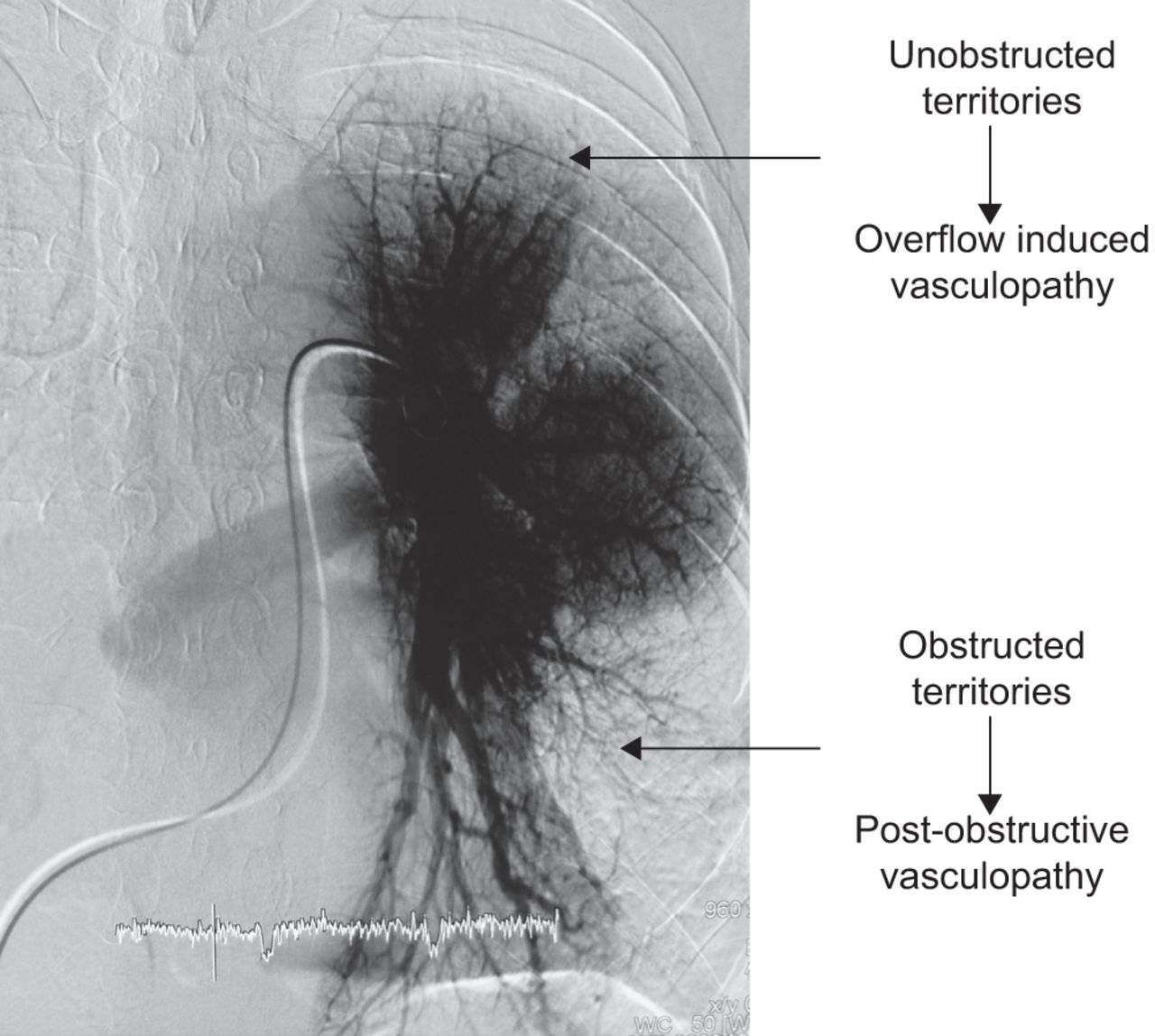
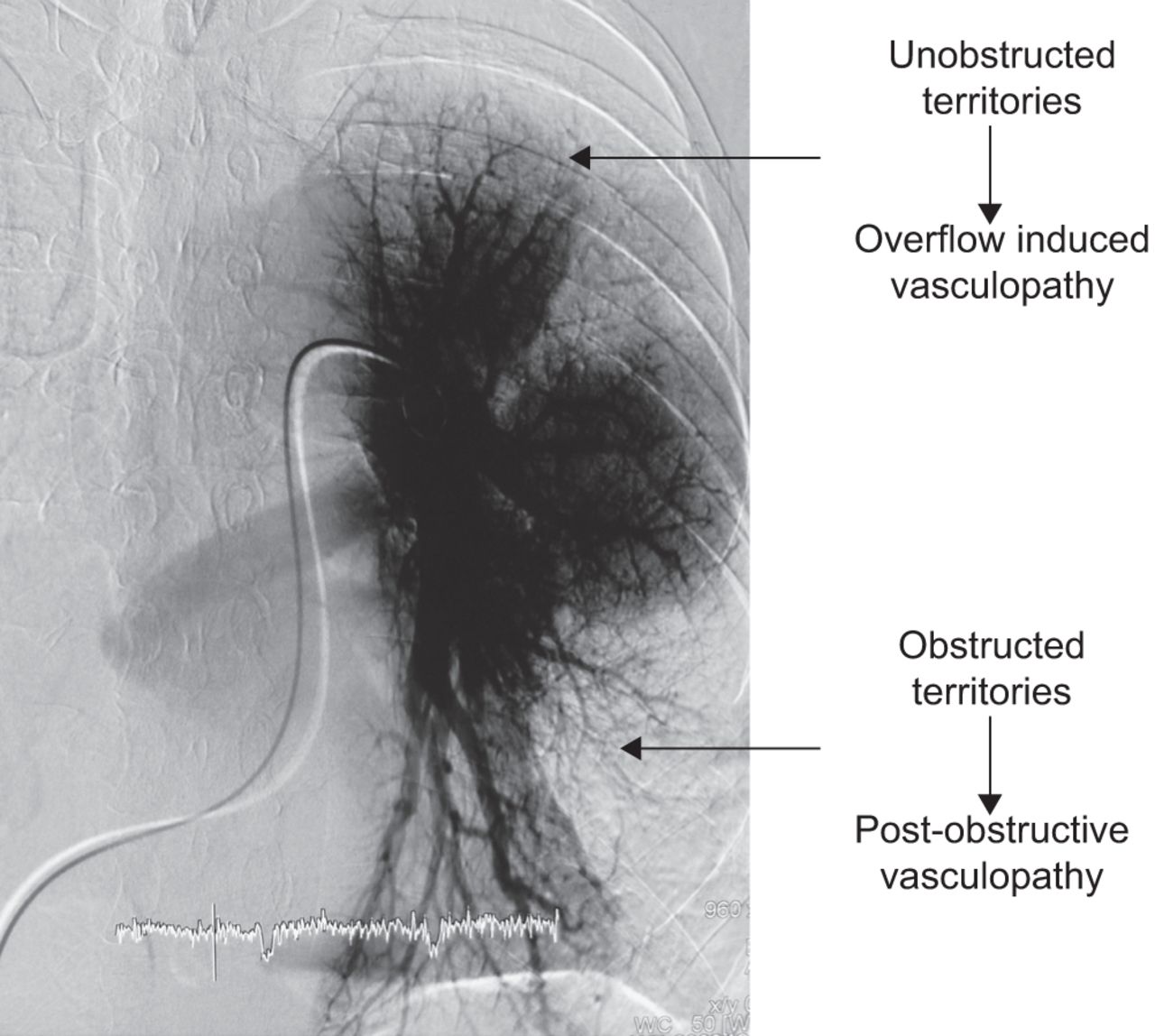
米奥泽et al。[4]首次描述了CTEPH患者肺血管床的两个腔室:一个是受慢性缺血影响的阻塞腔室,另一个是受流量和剪切应力增加影响的通畅腔室(图1).因此,除了大肺动脉梗阻外,CTEPH患者远端未梗阻区域也有血管病变。这些远端病变与其他形式PH患者的病变相似。
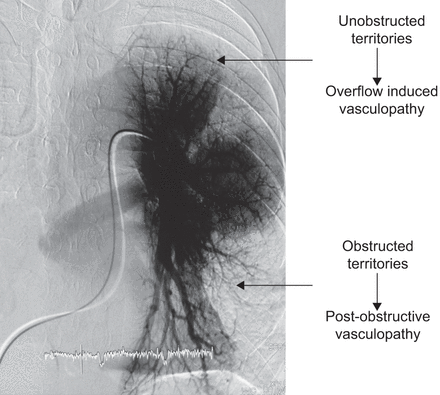
慢性血栓栓塞性肺动脉高压患者的前后肺血管造影显示两个血管区域。梗阻区出现慢性缺血,通畅区血流增加。
许多观察结果与这两个区域在增加肺血管阻力中的作用一致。首先,对于肺扫描测量的相同程度的肺动脉阻塞,与急性肺栓塞相比,CTEPH与更高的肺阻力值相关[46]。其次,CTEPH发病的特征是无症状的蜜月期,在此期间肺血管阻力逐渐增加,没有复发栓塞的证据。这两种血管腔室已分别在动物中复制,最近,在仔猪模型中同时复制。
复制肺动脉阻塞的模型
首次尝试在动物模型中复制慢性肺动脉阻塞是由F阿德尔et al。[24他们使用近端栓塞线圈和组织粘合剂进入左肺动脉。左肺动脉慢性梗阻(持续5周)导致无ph值的慢性肺缺血。其结果是肺系统血供增加通过支气管,纵隔,肋间动脉,以及远端阻塞性肺血管病变。这些病变与慢性肺动脉结扎术后的病变相似[25]。
选择左肺动脉结扎,因为其易于重新植入肺动脉主干,复制PEA后的再灌注损伤。对慢性肺血管阻塞的全身血管反应因物种而异,在大型动物(狗和仔猪)和大鼠中,支气管动脉增殖到肺壁内气道,在小鼠中,肋间动脉增殖到胸膜间隙[26]。虽然CTEPH研究主要在仔猪中进行,但在大鼠肺动脉结扎模型中进行了一些支气管循环研究[27]。阻塞其中一条肺动脉可刺激同侧肺的支气管血管生成[28]。肺动脉结扎后2-3天,支气管动脉开始扩张,供应肺循环通过前毛细管的吻合。这些吻合可维持气道上皮的氧合,从而解释了慢性肺缺血后再灌注损伤程度较急性肺缺血小的原因[29]。
除了支气管循环肥厚外,阻塞性血管病变的特征还包括肺动脉异常,包括中膜厚度增加[30.],血管反应性受损[31],内皮一氧化氮合酶功能受损[30.],增加对内皮素(ET -1)的反应性[32]导致再灌注后阻力增加[33]。
慢性缺血后肺再灌注后阻塞性血管病变逐渐逆转[25,34,早期缺血-再灌注损伤伴内皮细胞损伤后[35]。
主动脉-肺分流的关闭复制了PEA后通畅区域的血流动力学情况。分流关闭诱导ET-1和ETA表达正常化,随后远端肺动脉中膜肥厚逆转。这些结果与PEA后3-6个月肺血管阻力逐渐改善一致。
复制无阻塞肺动脉病变的模型
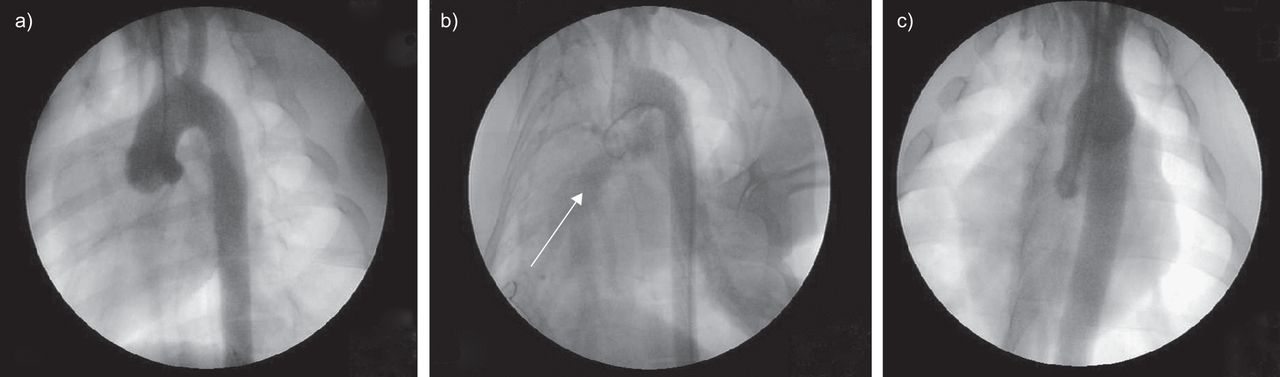
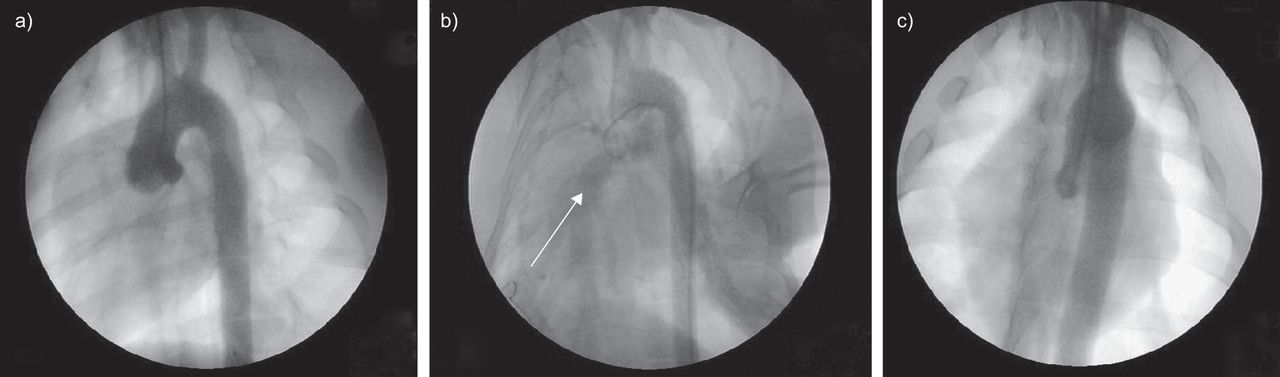
在CTEPH的早期阶段,由于心输出量的重新分配,通畅的区域受到慢性血流量增加的影响。因此,全身性-肺分流术已在动物中应用,以复制CETPH患者通畅区域所见的病变[36]。在最初由R恩达et al。[37为了复制先天性心脏病,通过在升主动脉和肺动脉主干之间植入短假体来诱导主动脉-肺分流。该模型已用于诱导高流量肺血管病变,类似于CTEPH中通畅区域所见的病变(图2).与左侧肺动脉结扎模型一样,通过胸骨正中切开术实现主动脉-肺分流,以避免胸膜打开和随后的炎症反应。5周的主动脉-肺分流术诱发高流量肺血管病变,其特征是远端肺动脉中膜厚度增加(图3)与平滑肌细胞增殖(如增殖细胞核抗原标记所示)和肺组织中ET-1及其受体内皮素受体A (ETA)水平升高有关。ET-1是一种有效的血管收缩剂和血管平滑肌细胞有丝分裂肽。ET-1过表达已在其他高肺流量动物模型中发现[38- - - - - -40]。ET-1分泌过剩可能是对动脉压升高引起的剪切应力等刺激的反应[47]。
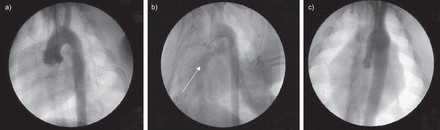
假组仔猪主动脉造影术。a)主动脉弓与两条主动脉上动脉。b)主动脉-肺分流术建立后,肺血管床(白色箭头)与主动脉弓同时可见。c)分流关闭后肺血管床未见混浊。
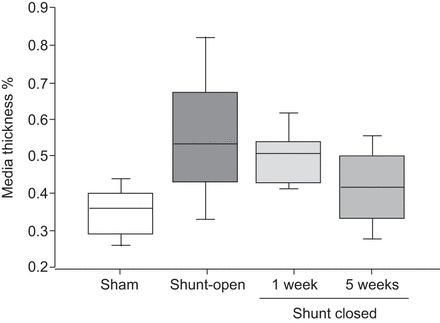
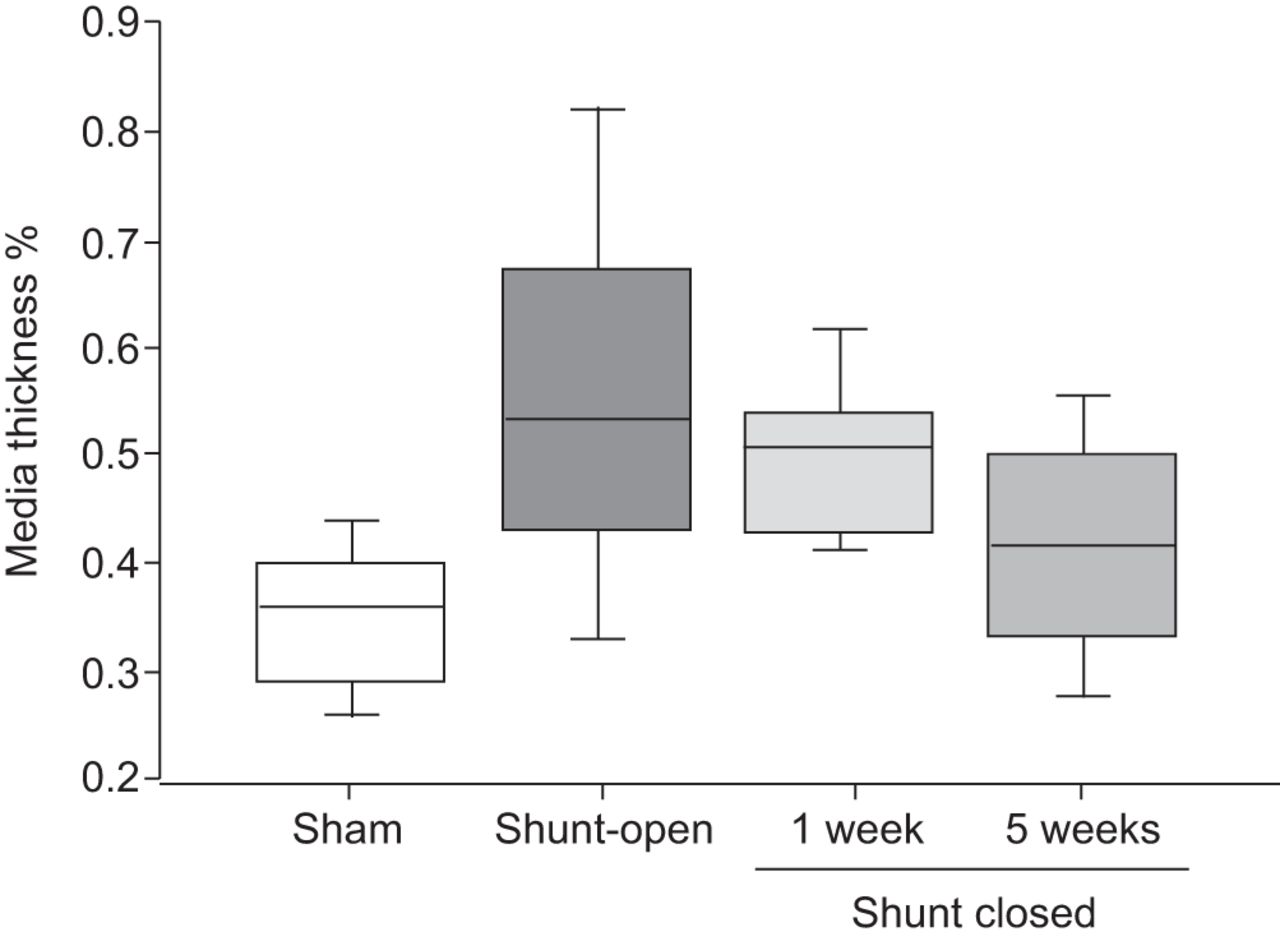
假手术组(35.9±0.8%)、分流开放组(55.6±1.2%)、分流封闭1周组(48.7±1%)、分流封闭5周组(40.9±1%)中肺介质厚度占肺小动脉(<150 μm)百分比的箱线图[36]。介质厚度百分比计算如下:外径减去内径,再除以外径。通过在仔猪升主动脉和肺动脉干之间的移植物插入,产生了主动脉-肺分流。分流的闭合是通过分割移植物来实现的。
复制cteph所有特征的动物模型
尽管前面描述的动物模型提供了关于血栓溶解受损以及阻塞和通畅区域病变病理生理学的有用信息,但它们未能复制人类CTEPH的重要特征,包括肺动脉高压、两个肺血管间室之间的相互作用,以及最重要的右心室重塑和功能障碍。
自20世纪90年代以来,多次尝试建立CTEPH动物模型[48- - - - - -52]由于非常有效的内源性纤溶系统溶解血块而失败[53以及肺循环显著的适应能力。因此,犬急性肺栓塞后3小时[49],只有30%的初始注射血栓体积留在肺动脉内。加入氨甲环酸[49]或纤溶酶原激活物抑制剂-1 [50延缓注射之间的血栓吸收未能解决这一问题。第二个困难是肺循环储备大,需要阻塞一半以上的肺血管才能增加肺血管阻力。肺血管系统的适应能力解释了为什么反复注射小的,惰性的,不可吸收的物质不能复制CTEPH。因此,在慢性肺动脉注射陶瓷珠的犬模型中,每次注射后1周内,直径为3mm的陶瓷珠,肺压和阻力恢复正常[51]。采用100 ~ 300 μm微球,反复栓塞60天,提高肺动脉压[52]。60天后开始出现PH征象,但未见支气管动脉肥大、后梗阻性或高流量血管病变、近端血管梗阻性[52]。第三个挑战在于复制房车的改造。广泛急性肺动脉树梗阻通常是致命的心力衰竭,以前没有右心室训练和肥厚。为了耐受肺动脉压的持续升高,右心室必须经历重构,这包括逐渐的心室壁肥厚,然后是右心室腔增大和矛盾的室间隔运动。在持续肺动脉高压的情况下,右心室衰竭。
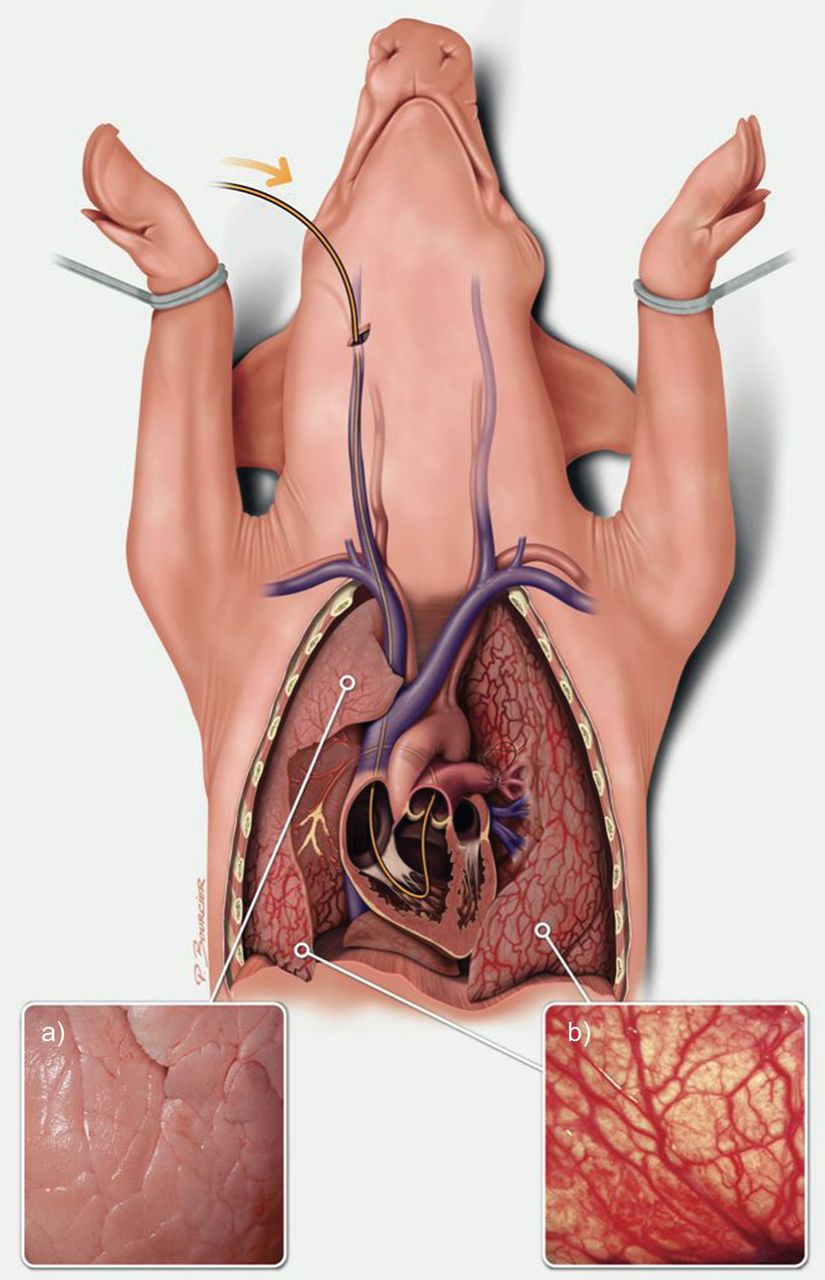
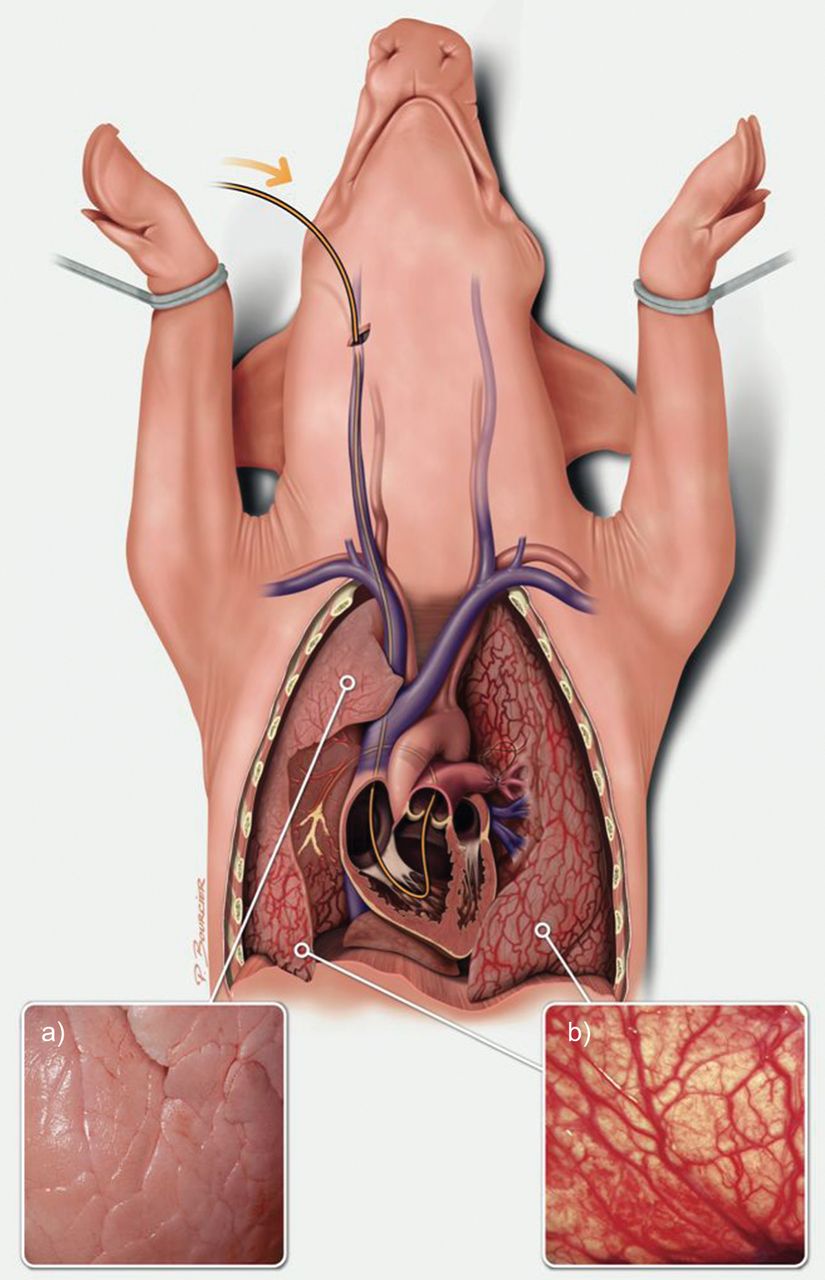
我们最近开发了一种CTEPH仔猪模型[41],包括原发性左肺动脉结扎术通过胸骨切开术后,在透视控制下,每周经导管栓塞,将组织粘合剂(histoacryyl)植入右下叶,持续5周。肺动脉结扎使肺循环储备不堪负荷,导致几周内PH值升高,剩余肺血管进行性梗阻。阻碍的进展性已经实现通过每周栓塞使右心室适应压力增加,从而防止急性右心室衰竭死亡。组织胶粘剂囊泡在接触血液后立即凝固并粘附在动脉壁上。结果是右下叶动脉内未解决的物质造成近端梗阻。因此,右上叶动脉保持通畅,并表现出与CTEPH中通畅区域相同的病变。5周后,该仔猪模型复制了人类CTEPH的所有特征:肺血管阻力增加,平均肺动脉压力增加,阻塞区和未阻塞区远端肺动脉中膜厚度增加,阻塞区支气管动脉全身性血供增加,右心室肥大,右心室增大和室间隔不规则运动(图4).有趣的是,尽管栓塞在5周后停止,但肺血管阻力的增加在最后一次栓塞后持续了1个月(未发表的数据)。ET-1及其受体ETA和ETB的过表达被记录在未阻塞区域重塑的远端动脉中,与先前在仔猪高流量血管病变中的发现一致[36,38]。此外,ETA在梗阻性血管病变中的过表达与既往研究的后梗阻性血管病变的结果一致[33]。需要进一步的实验来监测距离最后一次肺栓塞距离的肺阻力,以研究右心室重塑和衰竭,并评估阻塞区域和通畅区域之间的相互作用。然而,人们应该记住,该模型不能复制在人CTEPH中看到的受损的血栓溶解。
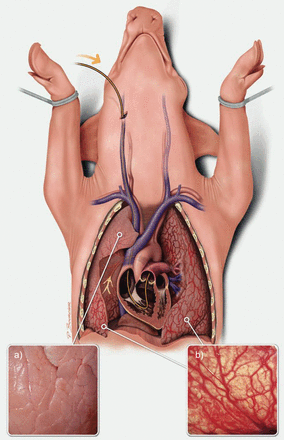
慢性血栓栓塞性肺动脉高压仔猪模型由左肺动脉结扎和右下叶反复栓塞组成:缺血肺区支气管动脉肥大(b),右上叶明显正常(a),由组织粘合剂和纤维蛋白制成的未溶解的血管内物质塑造动脉树,右心室重塑。
结论
几十年来开发的动物模型为CTEPH的病理生理学提供了有价值的信息。在几种动物模型中分别研究了血栓溶解和阻塞和通畅区域。最近,一种复制人类CTEPH所有主要方面的模型被开发出来。该模型对于研究右心室功能障碍和远端肺血管异常是有用的。然而,还需要更多的模型来阐明血栓持续存在的病理生物学。
脚注
本系列以前的文章。1号:Delcroix M, Vonk-Noordegraaf A, Fadel E,et al。慢性血栓栓塞性肺动脉高压的血管和右心室重构。呼吸呼吸J2013;41: 224 - 232。2号:Lang IM, Pesavento R, Bonderman D,et al。慢性血栓栓塞性肺动脉高压的危险因素和基本机制:目前的认识。呼吸呼吸J2013;41: 462 - 468。3号:Jenkins DP, Madani M, Mayer E,et al。慢性血栓栓塞性肺动脉高压的外科治疗。呼吸呼吸J2013;41: 735 - 742。4号:裴克-扎巴J,杨莎P,金NH,et al。慢性血栓栓塞性肺动脉高压:药物治疗的作用。呼吸呼吸J2013;41: 985 - 990。
权益声明书
没有宣布。
- 收到了2012年7月2日。
- 接受2012年12月8日。
- ©2013人队











{kind=link}
{kind=link}
{kind=link}
{kind=link}
![Box plot of media thickness as a percentage of small pulmonary arteries (<150 μm) calculated in: the sham (35.9±0.8%), shunt-open (55.6±1.2%), 1-week shunt-closed (48.7±1%), and 5-weeks shunt-closed (40.9±1%) groups [36]. Media thickness percentage was calculated as follows: external diameter minus internal diameter and divided by the external diameter. Aorto-pulmonary shunting was created by a graft interposition between the ascending aorta and the pulmonary trunk in piglets. Closure of the shunt was obtained by dividing the graft.](http://www.qdcxjkg.com/content/erj/41/5/1200/F3.large.jpg?width=800&height=600&carousel=1){kind=link}
{kind=link}
{kind=link}
{kind=link}