条文本

文摘
背景原发性纤毛运动障碍(PCD)基因异构条件丰富一些血亲的人群,结果从隐性突变影响纤毛生物起源和能动性。目前,诊断需要多个测试专家。
方法基因诊断实用程序面板下一代测序(上天)是在161年评估从多个人口家庭血统无关。
结果大多数(82%)家庭影响了个人与biallelic或半合(75%)或单(7%)致病因果基因等位基因在已知的纤毛。丧失等位基因占主导地位(73%转移,stop-gain拼接网站),大多数(58%)是纯合子,即使在non-consanguineous家庭。尽管57%(88)总155诊断疾病的变异是小说,复发性突变,突变基因检测。这些白色的欧洲之间明显不同(52%的家庭DNAH5或DNAH11突变)、阿拉伯(42%的家庭CCDC39或CCDC40突变)和南亚(单身LRRC6或CCDC103变异在36%的家庭)患者,按人口来揭示一个引人注目的遗传分层纤毛运动的起源。基因促进成功的诊断81%的家庭与正常或不确定的超微结构和67%失踪之前超微结构的结果。
结论这项研究显示了高通量的增值目标门店在加快纤毛诊断。因此,病人有潜在的重大利益早些时候在更广泛的和/或遗传筛查的实施。
- 原发性纤毛运动障碍
- 突变谱
- 纤毛
- 支气管扩张
- 人口
来自Altmetric.com的统计
介绍
原发性纤毛运动障碍(PCD)是一种罕见的遗传性疾病引起的纤毛蠕动障碍与一系列相关联的缺陷能动的纤毛结构和生物转化。纤毛运动通常是一个常染色体或X与隐性障碍引起的突变结构纤毛蛋白> 40个不同的基因编码,纤毛的组装和运输涉及multiciliogenesis因子和蛋白质。1 2儿童和成人影响PCD因此清单与进步的呼吸道疾病的特点是支气管扩张和肺功能受损。症状通常出现在早期生活与新生儿呼吸窘迫综合征与慢性持续湿咳嗽、鼻炎、鼻窦炎、中耳炎和听力缺陷。3大脑有缺陷的纤毛的室管膜、输卵管和胚胎发育可以解释其他疾病的功能。一半的患者有一侧缺陷引起的胚胎节点纤毛功能障碍,和很大一部分的男性患者与有缺陷的精子鞭毛临界正常值。受影响的个人,特别是那些有纤毛数量减少,还可以与脑积水表现,而RPGR和OFD1分别突变可以导致罕见的视网膜营养不良和金刚石亚型oral-facial-digital综合症。1 2 4 5
PCD的患病率大约是000年全球一点十五分。PCD发生更频繁地在英国等高度近亲社区南亚人口,在疾病患病率高达1:2300谁。3一般来说,纤毛运动症状变量和诊断是经常延误或错过了。6早期诊断有潜力提高发病率由于肺损伤可以推迟了专业护理。3个7PCD诊断检测需要访问调查的组合包括测量低鼻一氧化氮水平,高速视频显微镜纤毛击败缺陷,纤毛超微结构缺陷通过透射电子显微镜(TEM)分析,异常的能动的纤毛蛋白免疫荧光染色,越来越多的基因分析。7 8
纤毛运动基因诊断需要识别biallelic常染色体或半合X突变有关。7 8基因突变在已知纤毛被发现在60% - -70%的患者检测PCD。1 3额外的基因仍然被识别,基因检测的敏感性作为“黄金标准”减少了诊断测试。然而,随着进步的识别整个病态的基因组的金刚石和持续的减少导致DNA测序成本,遗传学可以越来越被视为一个一线测试诊断途径。基因板目前可以更有效的为目标序列覆盖率和减少时间和成本比全外显子组和基因组测序。9
在这里,我们提出一个针对下一代基因测序(上天)面板的方法描述multiancestry群患者PCD。我们的目的是探讨这种方法的实用工具金刚石,临床和遗传异质性条件,目前的诊断需要多个测试专家。8
材料和方法
主题
一百六十一个无关的家庭证实自我祖先和血缘关系的时候招聘从英国国家PCD确定诊断和管理服务(伦敦皇家主管布朗普顿医院地铁站,南安普顿大学医院,利兹一般医院,布拉德福德皇家医院,伯明翰儿童医院和莱斯特皇家医院)和临床合作中心在葡萄牙(医院·德·圣玛丽亚,Centro Hospitalar葡京北,《里斯本条约》),巴勒斯坦(Makassed医院,东耶路撒冷)和埃及亚历山大大学儿童医院,亚历山大。招聘发生在2015年1月和2017年2月。PCD的诊断之后欧洲呼吸协会的指导方针,188bet官网地址8使用各种方法根据临床中心,包括临床表现和正式的PCD的诊断测试的结果(鼻一氧化氮水平,通过TEM纤毛超微结构分析,纤毛击败模式和频率高速视频显微镜和免疫染色对特定的纤毛蛋白)。研究入选标准是基于临床怀疑金刚石和/或可用的纤毛超微结构的TEM分析。TEM资料无法获得在共有27个家庭中,都包括在这项研究基于其他临床标准显示纤毛运动。
有针对性的门店
基因组DNA从全血中提取或者唾液样本使用目标门店基因突变筛查面板包含所有已知的PCD (在线辅助表S1)和隔离地层变位基因,再加上两个迭代的一套更大的纤毛motility-associated候选基因。这些都是后整理大量文献搜索候选人的纤毛或可能参与确认,并从数据从以前的人类遗传学和纤毛运动生物模型研究。小组调查设计使用安捷伦SureDesign工具(安捷伦科技,圣克拉拉,加州,美国)来捕获所有编码区域和25个基点exon-intron边界(在线辅助表S1和S2)。捕获探针与潜在的低覆盖率丰富的地区。SureSelectQXT挥动目标浓缩设备(安捷伦科技)是用于图书馆准备后,制造商的协议。双末端测序(2×150个基点)都使用了NextSeq 500/550高输出2装备和NextSeq测序平台(美国加州Illumina公司)。多路复用的48个样品是相同的流动细胞/测序运行。测序数据处理使用自身的生物信息学管道在北东泰晤士河区域遗传服务。10变体被过滤意义产生变体列表每个病人的兴趣,符合预期轻微金刚石(< 1%)等位基因频率和常染色体或X隐性遗传模式有关。变异是优先考虑基于他们小ExAc数据库中的等位基因频率,111542个人Born-in-Bradford群英国南亚人12和艾尔mena数据库的基因变异在中东和北非的个人。13潜在的致病性是评估使用几个软件包括人类拼接仪,筛选,PolyPhen-2 MutationTaster和结合注释依赖消耗的分数。变体致病性得分是根据指导方针的美国大学医学遗传学和基因组学(在线辅助图S1),14使用完善的分类(或堆叠)系统预测致病的可能致病,变异和不确定的意义可能良性或良性变异。所有的细节感兴趣的变异基因记录数字在线辅助表S3。为所有受影响的个人,寻找大型插入/删除突变和基因拷贝数异变(使用ExomeDepth软件)分别进行。15
桑格测序
所有优先变体被证实在渊源者和隔离在可用的家人利用Sanger测序。测序数据使用SnapGene查看(美国芝加哥GSL生物技术)或Sequencher软件(基因编码公司,安阿伯市密歇根,美国)。
结果
针对门店收益率高的诊断输出与PCD multiancestry患者群
无关的渊源者从161家庭使用目标门店基因筛选面板分析,其次是桑格sequencing-based隔离分析来确认所有识别感兴趣的基因变异和决定他们的家族遗传模式。所有家庭影响了个人暗示临床表型,除了要么(1)纤毛超微结构的缺陷确认(97户);或(2)金刚石仍然是高度不确定的TEM结果,暗示家庭(37);或(3)没有诊断TEM分析金刚石仍然执行但临床上高度怀疑家庭(27)。归纳了它们的细节表1。PCD-consistent特性总27的家庭渊源者缺乏TEM数据总结在线辅助表S4。祖先和血缘关系的家庭是总结在线辅助表S5显示,46%(74年)是欧洲,22%(35)南亚,18%(29)阿拉伯和其他其他祖先。血缘关系曾在161年的29%,最高水平在阿拉伯(25岁,86%)和南亚(12 34%)的家庭。
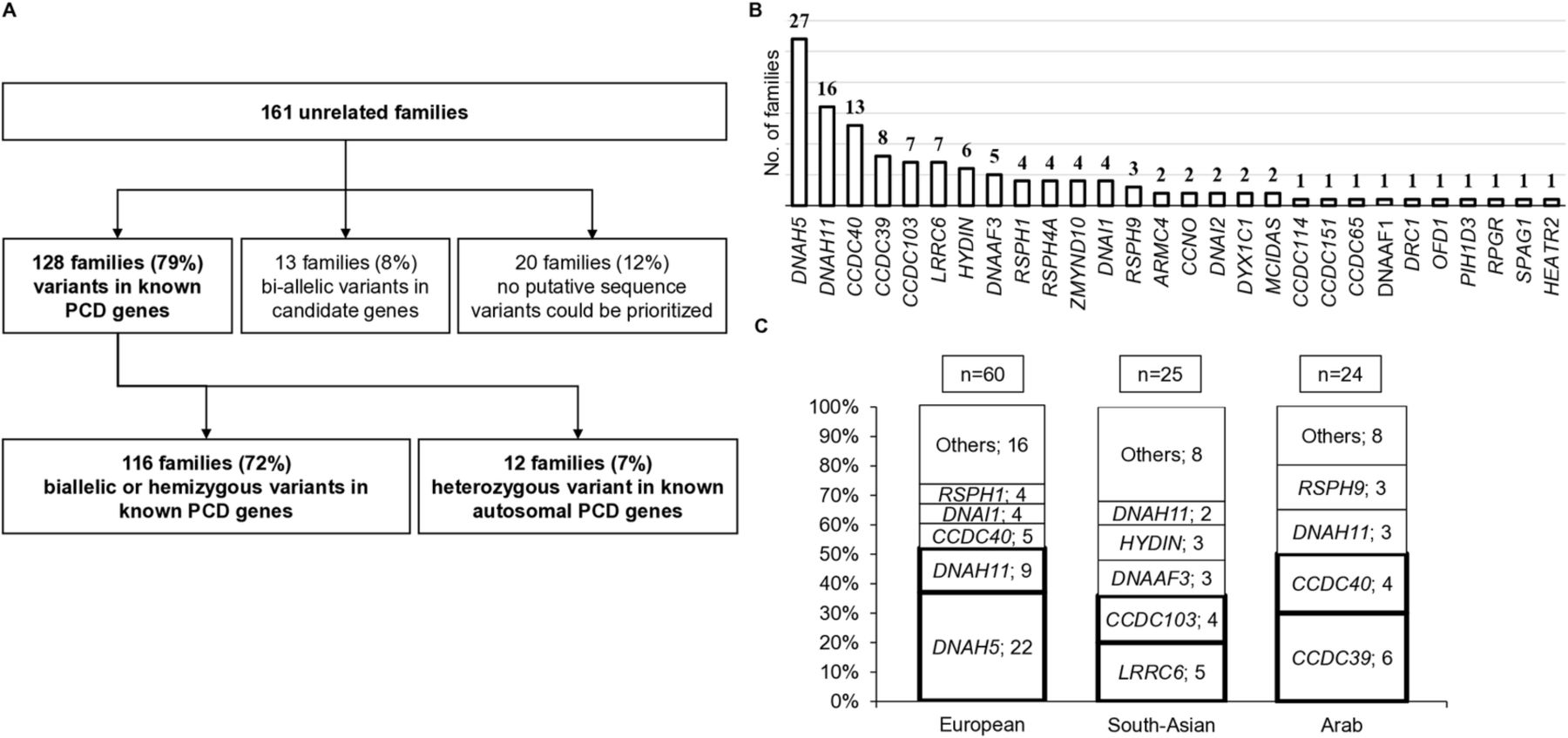
我们确定因果基因变异在已知纤毛金刚石的家庭在128年最初的161,随后4更由于候选人发表的纤毛运动基因,因此由最后82%的人群(图1一个和在线辅助表S3)。Biallelic常染色体变异有关的或半合X中确定116个家庭在已知纤毛基因,进一步组成的4科2CFAP300变体16和2DNAH9变体。17因为这些基因作为PCD-causing最近证实,这意味着120的161(75%)的家庭被诊断。在12个(7%)的家庭,只有一个突变等位基因(单杂合的)金刚石基因被发现在一个已知的常染色体,这被认为是一个不完整的基因诊断。然而,我们包括变体数据全部12个家庭,因为七蛋白质截断变异和五已经在先前的研究报道患者纤毛运动;此外,所有这些“单一打击”variant-carrying患者纤毛超微结构的缺陷符合涉及突变基因(图1一个和在线辅助表S3)。

针对下一代测序收益率高(72%)患者的诊断输出PCD和揭示了一个不同的突变景观由祖先分层。(A)流程图的遗传结果发现家庭参加这项研究描述了基因诊断输出。渊源者从161年无关家庭受到下一代测序基因能动的使用ciliome面板。影响个人在4 13“候选基因”的家庭进行biallelic现在在两个基因变异被认为是致病,CFAP300和DNAH9;详见正文,这增加了诊断收益率为82%,而不是79%。(B)的遗传分层multiancestry队列的所有161个家庭发现携带基因突变在已知的纤毛。(C)的突变基因在三个不同的数量说明不同的祖先有不同的基因。在109年欧洲、阿拉伯语和南亚的家庭,发现最常见的基因突变DNAH5在欧洲家庭(37%),LRRC6和CCDC103在南亚的家庭(36%)CCDC39和CCDC40在阿拉伯家庭(42%)。PCD、原发性纤毛运动障碍。
13(8%)的家庭,biallelic金刚石(在候选基因变异CFAP300,DNAH9包括确定),并进一步这些基因的功能描述和他们的角色在导致纤毛正在进行。最后,20(12%)的家庭没有公认的重要的序列变异,其中11有纤毛超微结构的缺陷被TEM和3有低鼻一氧化氮和纤毛异常拍频但不确定TEM (在线辅助图S26),另一个拥有强大的临床怀疑金刚石(内脏转位和复发性呼吸道疾病)的事先调查。
重要,基于遗传分层构成PCD
优先考虑变异为128年诊断家庭被确定在不同功能类别的PCD基因(图1 b)。最普遍的,在38%的家庭,影响基因编码外动力蛋白臂(ODA)组件(DNAH5,DNAH11,DNAI1,DNAI2)。第二个集体最常见影响基因编码动力蛋白装配因素(LRRC6,DNAAF3,ZMYND10,DYX1C1,DNAAF1,PIH1D3,SPAG1,HEATR2)≈17%的家庭,其次是统治者蛋白质的突变基因(CCDC39,CCDC40)在16%和径向辐条(RSPH1,RSPH3,RSPH4A,RSPH9在8%的家庭)。CCDC103突变影响5%的家庭,但在其他方面的突变基因在ODA对接(ARMC4,CCDC114,CCDC151),中央对HYDIN)和nexin-dynein监管复杂结构(CCDC65,DRC1),multiciliogenesis (CCNO,MCIDAS),或造成“综合征”的形式(OFD1,RPGR)更罕见,影响集体≈9%的家庭。
总的来说,DNAH5是最常见的基因突变,突变中确定影响个人来自27个(21%)的家庭(图1 b和详细的图2一个)。然而,人口不同的祖先(种族)有相当不同的遗传资料。DNAH5和DNAH11突变被发现在37%和15%的欧洲家庭,分别,但只有少数的病人从其他祖先。LRRC6和CCDC103最常见的突变基因在南亚的家庭,影响整体超过三分之一(分别为20%和16%)的家庭。CCDC39和CCDC40是主要的两种突变基因影响阿拉伯人口,发现42%的阿拉伯家庭(图1 c)。如下详细的进一步,这些频率是由于周期性的混合物,假定奠基者效应突变,突变往往独特的个体家庭。

PCD-causing突变分布multiancestry队列是由蛋白质删除和纯合突变。(A)的示意图DNAH5突变确定在这项研究中,如果之前报道的用黄色标记。复发性突变是盒装粗体。保守域的DNAH5蛋白质基因组结构表示。根据(NM_001369.2)转录变异屈指可数。(B)家庭基因突变在已知纤毛分组根据接合性状态显示,大约58%的突变确定在本研究中患者在纯合状态。(C)突变分类根据各自的蛋白质的影响表明,转移和无意义突变是最普遍(58%)、错义变化剪接缺陷为15%和21%。集体,突变预测蛋白质删除影响约占77%(转移,胡说八道,剪接缺陷和CNV)。(D)中确定突变biallelic状态在常染色体基因或半合状态X相关基因的分类根据指导方针从美国大学医学遗传学显示82%的突变类5(显然致病性),8%类4(3班可能致病)和10%(不确定的意义)。PCD、原发性纤毛运动障碍。
扩大基因突变谱在已知的纤毛
很大一部分的家庭(128年74年,58%),从所有祖先群体,被发现携带变异纯合子(图2 b)。令人惊讶的是,有三分之一的欧洲家庭60(20),主要考虑non-consanguineous,携带变异纯合子在已知纤毛基因(在线辅助图S3),强调可能未被发现的同系交配和关联性。Biallelic导致常染色体基因杂合变异体疾病31%的患者家庭(39),和3家庭半合变异被发现在X相关基因(PIH1D3,RPGR,OFD1)。
全部167个变异的PCD基因检测到128个家庭,主要变体类型预测蛋白质删除(73%)列为移码突变(32%)、无稽之谈(26%)和突变影响剪接(15%)。错义突变占21%的变异。基因拷贝数异变和inframe删除或删除/插入突变占总体变异的6% (图2 c)。12个单变体识别没有第二个突变(“一打”患者在线辅助表S3)没有被认为是诊断,但在诊断116个家庭的155个变异(不包括DNAH9和CFAP300等位基因)82%是致病性(5类)和8%是可能的致病(第4类)。3班临床意义不明的变体(VUS开头)代表只有10%的变异;这些在一些警告提供明确的诊断(图2 d)。
突变的频率和频谱之间的显著区别不同的祖先
穿过人群,除了许多family-unique突变的存在,大量的流行复发性突变,推测反映人口瓶颈/创始人的影响,发挥着重大贡献不同的突变基因影响金刚石数量不同。我们定义14反复突变共同占大量PCD-causing变体。有些ancestry-specific和其他人出现在多个种群(表2)。在60欧洲家庭因果等位基因被定义,三个复发DNAH5突变被发现占13%(120年16日)欧洲疾病等位基因的两个创始人尽可能之前报道的影响(c.10815delT;p。Pro3606Hisfs*22 and c.13458_13459insT; p.Asn4487fs*1)18和一个无义突变(c.6261T > G;p.Tyr2087 *)不是之前报道(表2,在线辅助表S3和图2一个)。其他三个之前报道突变一起占13%(120)16日欧洲疾病等位基因:DNAI1c.48 + 2 dupT;p.Ser17Valfs * 12,RSPH1c.275-2A > C;p。Gly92Alafs*10, and a recurrent homozygousDYX1C13.5 kb基因删除。19日至22日DNAH11突变金刚石疾病欧洲也是一个主要因素,但主要是family-unique而不是周期性的等位基因。
在南亚的家庭中,一个先前描述LRRC6突变(c.630delG;p.Trp210Cysfs * 12)是最常见的突变等位基因纯合子中发现五南亚的家庭地位。23先前报道CCDC103中检测出突变纯合状态(c.383dupG;p.Pro129Serfs * 25)在两个无关的南亚的家庭。24在一起,这两个变异仅占28%(50)14日所有疾病的等位基因在因果等位基因的家庭在25个南亚定义(表2和在线辅助表S3)。一个已知的复发性阿拉伯贝都因人RSPH9突变(c.801_803delGAA;p.Lys268del)中检测出纯合状态在两个阿拉伯家庭。19另一个可能的阿拉伯语创始人突变纯合子(c.1871_1872delTA;p.Ile624Lysfs * 3)CCDC39被发现在四个巴勒斯坦家庭。在一起,这两个变异占29%(48)12所有疾病等位基因的24阿拉伯家庭因果等位基因在哪里定义(表2和在线辅助表S3)。CCDC40金刚石疾病突变也为阿拉伯语,但family-unique的形式而非复发性等位基因。
其他复发性变异,之前报道的常见的南亚CCDC103错义突变(c.461A > C;p.His154Pro)检测主要在南亚人,而且在欧洲和其他祖先。25我们还发现了两个反复在不同的血统CCDC40突变,一个之前报道(c.248delC;p.Ala83Valfs * 84)20 21和一本小说(c.2824_2825insCTGT;p.Arg942Thrfs * 57),除了一个小说DNAH11突变(c.13494_13500del;p.Ser4498Argfs * 15)。
针对门店显示同义变异预测影响拼接是纤毛运动的一个原因
我们确定了两个同义的编码区变异不改变编码蛋白质的氨基酸序列预测,但预测影响剪接。一个,在一个阿拉伯家庭(PCD-G086)纤毛微管杂乱无章和内在动力蛋白臂(IDA)损失,是一个CCDC40纯合子的变体(c.48A > G;p.Gly16Gly)正确隔离在大家庭(在线补充S4数据和S5)。另一个,在一个欧洲的家庭(PCD-G093)纤毛ODA损失,是一个DNAH5同义突变(c.5157C > T;p.Phe1719Phe)加上一个错义变体(c.10815T > G;p.Asp3605Glu) (在线辅助数据S6和S7)。虽然有可能这些同义变异可能影响剪接,他们正在执行3班的vu (在线辅助表S3不能重新分类)和致病性或可能致病没有进一步工作提供直接观察他们的假定的拼接效果。
有针对性的门店是一个强大的工具来诊断和描述患者的PCD
TEM分析检测超微结构的缺陷97年134(72%)的家庭。其他37个正常TEM(如联系在一起DNAH11和HYDIN缺陷26日27日)或少数的不确定的TEM结果(表1)。最常见的超微结构的缺陷是ODA损失(61年45%,家庭),单独(23%,31个家庭)或结合IDA损失(22%,30家庭)。其他缺陷包括微管杂乱无章,有或没有IDA损失(12%)、中央微管复杂缺陷(6%),主要分离IDA损失(4%)或缺乏纤毛(5%)(表1)。
我们证实了很强的相关性在整个队列之间的基因缺陷和预期的超微结构的缺陷,在协议与纤毛文学(表1和在线辅助表S1)。2因此,TEM缺陷可以有价值的解释基因测试结果;然而,该研究还表明,他们并不总是必需的。161年的27(17%)的家庭没有TEM数据,仍然具有较强的临床怀疑,18在已知PCD biallelic变异的基因;因此,大部分(67%)被遗传学没有自信地解决了TEM信息(在线辅助图S8)。作为一个警示,少数患者(n = 6,星号在线辅助表S3)没有记录TEM缺陷纤毛运动状态的确认,他们还带纯合子或biallelic杂合变异体是罕见的,在已知的纤毛运动基因,但执行3班的vu不确定的意义。例如PCD-G013 biallelic杂合的两个DNAH5错义变化这两个未知的意义(不是之前报道)。在这些情况下,三个有变异HYDIN和DNAH11相关基因与正常TEM (PCD-G104, PCD-G017 PCD-G021),但对于其他的TEM可以检查和重复。
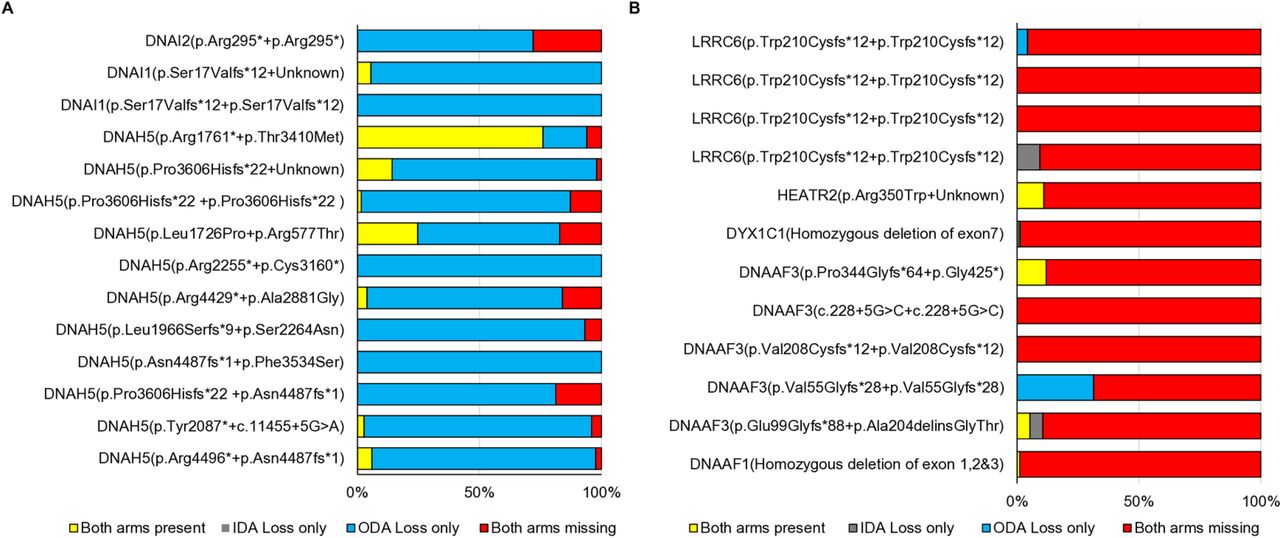
进一步测试的基因检测诊断工作流程的纤毛运动,我们详细看特定PCD的相关基因突变与纤毛超微结构的缺陷由TEM单一诊断中心。我们发现ODA基因的突变DNAH5与(1)明确的有关ODA像预期的损失,但有时也可以记录(2)结合艾达+ ODA或损失(3)不确定的TEM分析(图3一,表1)。类似的分类是可能的在个人动力蛋白组装基因突变(LRRC6,HEATR2,DYX1C1,DNAAF3或DNAAF1),结合艾达+ ODA预计损失(图3 b,表1)。通过观察基因突变的TEM上下文中的数据,我们可以定义一个不同的模式和区别这两个基因的功能类别,自动力蛋白组装突变导致结合艾达+ ODA损失大部分纤毛横截面,对比DNAH5突变导致主要ODA的损失。因此,基因数据允许这两个类别的基因诊断病人是杰出的(图3)。

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
金刚石和克服其他遗传学可以更好地描述患者诊断检测不一致。对于一组选定的患者,纤毛的百分比横截面显示亏损或内外动力蛋白臂都被记录在常规透射电镜诊断皇家主管布朗普顿医院地铁站设置参考潜在的遗传缺陷。(一)患者携带biallelic突变ODA组件显示主要是一个孤立的ODA的损失。结合艾达+ ODA(内在和外在动力蛋白臂)损失也会被记录下来。有趣的是相当数量可变的横截面显示正常的纤毛超微结构。(B)患者携带biallelic动力蛋白装配的突变基因显示结合失去双臂在大多数横截面。变量分离臂损失也报道。特战分队,外动力蛋白臂;PCD、原发性纤毛运动障碍;TEM,透射电子显微镜。
讨论
有潜在疾病异质性高,没有黄金标准测试可用来排除纤毛运动,所以测试的组合解释的临床症状往往是用于诊断。这越来越多的包括遗传分析。8这里,遗传筛查的效用评价群体的161金刚石家庭不同的血统无关,包括欧洲、阿拉伯和南亚,通过挥动筛选与额外的CNV的分析金刚石基因和已知的其他候选基因。这给了一个高收益的确认金刚石诊断或高度建议在75%的家庭。还有7%的家庭单杂合变异体在已知纤毛基因似乎因果可能可能携带第二次突变,门店无法检测,例如深intronic变体。
通过TEM分析明确的纤毛超微结构缺陷的识别仍然是一个金刚石诊断工作流程确认步骤,虽然不排除PCD未能识别TEM缺陷。8这里,我们确定基因突变在已知纤毛在81%的患者正常或不确定的TEM结果,意味着巨大的潜力结合遗传学诊断管道内的早些时候,正如前面所讨论的那样。27我们也可以诊断67%的患者具有强烈历史TEM并不可用,以及其他困难的情况下,例如,CCDC103p。H我s154Pro mutations, where other tests often give equivocal results.25
以前我们诊断输出高于大多数门店目标面板屏幕中,22 - 30类似于76%诊断成功实现从全外显子组测序(韦斯)和有针对性的CNV分析52个人。31日32缺席的局限性包括未知基因的面板,不确定意义的不完整的基因诊断当变异或金刚石基因检测单杂合变异体,技术问题影响序列覆盖深度,已知的生物信息学挑战识别基因拷贝数异变,33并与识别的一个众所周知的问题HYDIN突变,由于HYDIN2复制基因。26
本研究扩展了金刚石通过识别的遗传景观和突变谱61之前报道和88年以前未被发现的变异;因此,57%的变异机密在这里可能致病小说(在线辅助表S3)。大多数是蛋白质删除突变,预测功能丧失的等位基因,与先前的报道一致。同义突变一般是不报道中,但我们确定了两个预测导致拼接的变更,提高的重要性,寻找潜在的同义变体在尚未解决的情况下。
与之前的研究结果相一致,大多数发现金刚石变体私有的1 8;然而,一些被发现在多个不相关的家庭往往是更频繁的在特定的人群。有趣的是,有三分之一的欧洲家庭纯合突变尽管欧洲血亲记录率低,只有一个欧洲家庭报告血缘婚姻。在14个欧洲家庭,他们发现突变在文献中报道过的,暗示他们可能反映了欧洲创始人的影响。总的来说,我们发现DNAH5是最常见的金刚石协议与其他研究中的基因突变,34但这并不是在所有的祖先。我们确定了DNAH5变异在37%的欧洲家庭,代表欧洲人最常见的基因突变。相比之下,LRRC6和CCDC103在南亚突变更普遍的家庭,其中我们发现只有一个家庭DNAH5突变。在阿拉伯家庭,DNAH5突变被发现只有两个家庭CCDC39,CCDC40和RSPH9突变更普遍。
因此,研究发现了一个惊人的以人群为基础的基因分层底层纤毛运动。它突出祖先遗传的纤毛运动的影响,包括病人的重要性不同祖先中的说明PCD的完整基因景观。这信息是诊断相关,因为它可以用于改善,更小、更便宜的载体筛选面板针对特定人群和初步allelic-specific Sanger测序基因诊断,尤其是在门店设施的国家并不普及。遗传性疾病的临床相关性分层仍然知之甚少,但更多的研究新兴PCD genotype-phenotype相关性可以越来越多地影响疾病管理。17,第35 - 37 4
虽然我们可以确定基因型和纤毛超微结构的表现型之间的相关性好,一些差异在TEM分析结果明显即使相同基因的突变。DNAH5突变与ODA有关损失在大多数情况下,还有一些不确定的TEM结果的情况下,可能由于TEM在评估IDA的困难,3 8以及偶尔的录音结合艾达+ ODA损失往往是与动力蛋白组装基因突变。量化的胳膊带来损失的百分比DNAH5突变与基因突变在动力蛋白组装与遗传学表明增加TEM数据可以明显区分这两个组,DNAH5突变更高度与ODA损失和动力蛋白组装突变更高度结合艾达和ODA的损失。
总之,目标基因小组排序是一个具有成本效益的,省时间单独的测试在这个studyconfidently诊断至少75%的纤毛运动情况。它提高了诊断工作流程结果,证实纤毛运动不确定的TEM结果和帮助患者在TEM分析病人的诊断是不可用的。基因测试的灵敏度(诊断)金刚石与基因发现进步将继续增加。CFAP300,DNAH9,GAS2L2,LRRC56,MNS1,DNAH1和DNAH6都是基因已成为与PCD金刚石基因或确认为临时研究期间。16 17 38-42尽管目前金刚石基因列表不完整,这强烈支持的重要性,包括遗传学诊断途径进入,它可以发挥关键作用,克服的缺陷其他诊断措施。这可能是特别相关的国家访问其他专业PCD测试是不可用的。重大影响的基因和复发性突变出现在这项研究中,除了著名的祖先的遗传变异性纤毛运动的影响,对改进的分层的纤毛运动,以帮助促进更好地针对患者的诊断和疾病管理。
确认
我们非常感谢家庭参与本研究,感谢英国PCD家庭支持集团的继续的支持。我们感谢所有的实验室和临床成员金刚石在超高频诊断服务的国家,RBH和LRI促成了PCD诊断检测。我们感谢安德鲁·贾曼塞西莉亚Lo和金刚石基因Sudipto罗伊正式出版前的候选人名单。我们感谢丽莎·罗伯逊博士,临床遗传学服务,阿伯丁皇家医院NHS格兰扁,她的临床干预。
引用
补充材料
-
补充数据
仅这个web文件已经由英国医学杂志出版集团从一个电子文件提供的作者(年代)和没有对内容进行编辑。
脚注
推特@MahmoudFassad
调整通知这篇文章已经发表以来第一次修正。所属22已纠正。
贡献者MRF, MP, TC, JH, LJ, DJM-R,小王,嗯嗯的遗传和生物信息学分析和分析了遗传学数据。医学博士,AVR, CJ, PG, RH, AR, JT,美联社和SL参与生成和分析临床功能测试。,MD、AVR、PG AR和美联社纤毛超微结构进行分析。磁流变液、PA、EM、PC、COC RW, SC, WW,嗯,WS, LP CC, ML, EMKC, PK, NR、NF, JSL, CH和嗯确定病人和病人获得数据和样本。嗯的构思和监督研究和数据解释。MRF解释遗传学数据,开发了手稿草案和生成数据。磁流变液和嗯写手稿评论输入,TC,小王,JSL和CH。所有作者进行审核和批准最终的手稿。
资金全国PCD诊断服务是由英国国民健康保险制度。在南安普顿的研究支持NIHR南安普顿呼吸生物医学研究单位,NIHR威康信托基金会临床研究设施和AAIR慈善(Reg没有1129698)。本研究经费是由行动医学研究(GN2101;嗯)和大奥蒙德街儿童慈善拨款(V4515;嗯)和领导奖(V1299 V2217;嗯)。我们承认NIHR生物医学研究中心的支持大奥蒙德街儿童医院NHS信托基金会和伦敦大学学院(博士实习支持奖;磁流变液)。磁流变液是由英国文化协会Newton-Mosharafa基金和中国高等教育在埃及。工作,是一个独立的研究由国家卫生研究所的博士后研究奖学金和健康教育。 The views expressed in this publication are those of the authors and not necessarily those of the NHS, the National Institute for Health Research or the Department of Health. The authors participate in and acknowledge financial support from the COST Action BEAT-PCD: Better Evidence to Advance Therapeutic options for PCD network (BM1407), in particular two STSM grants awarded to MRF.
相互竞争的利益没有宣布。
病人同意出版不是必需的。
伦理批准患者发生招聘通知,适龄同意London-Bloomsbury研究伦理委员会批准(08年/ H0713/82)和合作机构的委员会。
出处和同行评议不是委托;外部同行评议。
数据可用性声明所有数据都包含在相关研究文章或作为补充信息上传。合理的请求数据。
作者注作者是成本的成员行动BEAT-PCD (BM1407)。