

文摘
氧气是救生和有毒。适当使用氧气的目的是提供一个两者之间的平衡效果。尽管当地氧中毒肺很接受,最近的证据质疑hyperoxemia在其他器官的床上的负面影响。心脏骤停后氧过多、创伤性脑损伤和中风已被证明会恶化的结果。hyperoxemia在机械通气病人的角色,面对无毒的氧气浓度的启发,尚不明朗。本文将审查的数据支持和反对使用保守的氧气目标和避免hyperoxemia机械通风病人。
介绍
几年前在一个RespiratoryC是期刊会议上氧气,1名誉主编大卫·皮尔森博士打趣说,“氧中毒是大脚怪,每个人都有听说过,但是没有人真正见过它”。氧过多是否相关临床关心的问题也存在一些问题。这些包括组织损伤及相关炎症过程的复杂性,治疗干预措施的影响,inter-individual遗传变异性。此外,问题是影响氧过多的争论是如何在过去的60年。历史背景继续颜色当代关于氧过多的临床重要性的看法和态度。即使是现在,当兴趣ventilator-induced肺损伤(VILI)占主导地位的讨论机械通风,氧过多的贡献的作用仍被认为是次要的。2在这篇文章中,我们双方的辩论这个问题。箴参数包含了背景信息必要欣赏主题的复杂性。
箴论点:氧疗法应该严格监管,以避免氧过多
肺氧中毒,现在被称为氧急性肺损伤,一直以来,大量的动物模型复制1783年安东尼·拉瓦锡的实验。1在本质上,暴露在FIO2≥0.70几天导致进行性肺损伤;症状和肺部病变的严重程度取决于浓度和持续时间。呼吸FIO2大约3 - 6 d≥0.80大多数动物通常是致命的。但是,似乎也有明显的种间甚至亚种的不同氧过多的炎症反应。氧急性肺损伤较小的物种几乎一致是致命的(如小鼠、大鼠、豚鼠、兔子),而人类出现更强的抵抗力,倾向于适应一段时间后约7 - 10 d通过肺泡ⅱ型细胞的增生。1尽管如此,超过230年的数据表明,人类肺部呼吸氧混合气体是有毒的,还有若隐若现的截然不同的可能性,氧过多可能是致命的患者具有遗传倾向。3
历史背景
医学兴趣氧过多导致的交集几个事件在20世纪中期,即军事需求与高空航空和潜水在二战期间和可用性的增加O2疗法治疗心肺疾病。从1950年代开始,第一个氧急性肺损伤的报道在人类医学文献中开始出现。4,5然而,就在1960年代,ICU的建立和长期机械通气以及高压O2临床治疗,严重的担忧2毒性反应了许多成年人和新生儿病例报告。1
然而,这些早期的报道暗示氧过多严重夸大了这个问题,几乎忽略了超过一个世纪的动物研究。开发或延续的过度归因急性呼吸衰竭氧过多的部分原因是知识的普遍缺乏对ARDS和VILI的局限性O2月初交付机械通风。只是发表的报告描述ARDS6以及其他研究ARDS的病理生理学,7- - - - - -9与呼吸道护理设备、相关的技术问题10- - - - - -12和VILI的说明13- - - - - -15氧过多的角色是置于一个更为现实的观点。不幸的是,当误解隐身特定现象消除,反应过度往往发生在相反的方向,培育一种过度怀疑的态度。在这一背景下当前的辩论必须陷害。在2016年的首要问题是如何在多大程度上,在哪个环境中氧急性肺损伤(,因此,需要严格控制FIO2)在关键疾病发病率和死亡率的影响。
进化和遗传影响对氧过多的反应
氧过多的影响对病人的结果,尤其是在ARDS,很难分离的复杂的相互作用机制,激活相同的损伤反应通路。最新进展在我们理解炎症和遗传学的作用表明,氧过多带来的问题超越了失衡活性氧(ROS)生产和抗氧化防御机制。相反,氧过多充当外部压力,强烈影响genetic-environmental交互。一个发人深省的例子就是在围产期暴露于氧过多生活深刻影响日后的几种疾病的发展,被认为是由后续的基因表达变化,曝光。16
生物进化与行星的崛起O交织在一起2浓度的波动周期与极端∼10到35%。17,18这对进化的生命形式的压力设计自适应策略成功地维持有氧代谢。对当前的争论源于这一事实爆炸在哺乳动物进化始于大约2亿年前。在此期间(三叠纪-侏罗纪大),大气啊2浓度下降大约35%至10%,因此选择性压力基因组中编码支持动物有效的呼吸系统。这一时期的特点是什么在真核生物进化是一个通过hypoxic-responsive调整代谢基因表达增强能力。另一个(前)进化特征是隔离代谢功能(氧化还原)从细胞DNA,防止氧化应激和遗传损伤。19
因此,应对缺氧,而不是氧过多,自然选择偏爱。但这引发了问题的效率共赢抗氧化防御机制在哺乳动物的进化旅程开始呼吸一个启发O2张力约为70 - 100毫米汞柱。此外,许多祖先物种主要居住在地下的栖息地可能增强他们适应低氧环境的能力。20.很可能这些基因编码策略影响所有当前生物反应的氧过多的方式我们还没有完全理解。
概述:细胞信号VILI和活性氧的作用
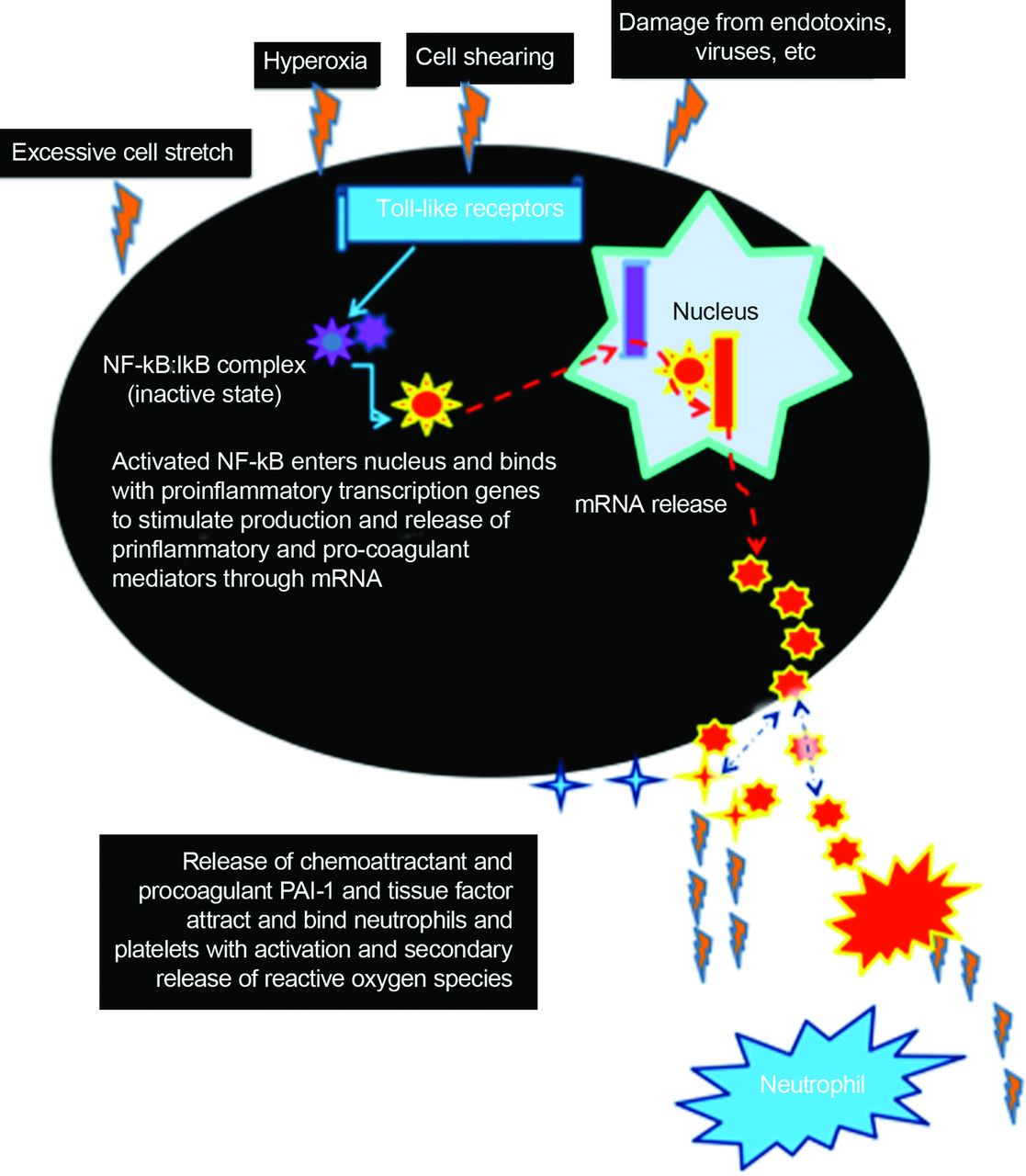
在VILI方面,目前的想法是,先天免疫系统起着关键作用的肺部炎症的发生和发展无论初始的事件。21被称为有关分子模式,它们代表一线细胞防御病原体和机械和化学压力。这些包含一个大家庭的细胞内分子称为toll样受体在不同刺激反应,包括细胞死亡(坏死、凋亡),免疫细胞激活,和碎片从基底膜的破裂释放出来。21反过来,toll样受体激活细胞内的核因子k B,至关重要的分子,激活多种基因参与细胞的防御机制。22
因此,核因子k B可以激活众多刺激呈现在至关重要的疾病,包括活性氧、促炎细胞因子,木糖醇,病毒,和stretch-related肺损伤。22,23有趣的是,核转录因子k B是抑制的抗氧化剂。22此外,核因子k B刺激stretch-related损伤和氧过多导致纤溶酶原激活物的释放inhibitor-1和组织因素通过气道和肺泡上皮细胞,内皮细胞,巨噬细胞和成纤维细胞。24,25纤溶酶原激活物的刺激inhibitor-1和组织因素诱发凝血级联,导致肺泡和小气道纤维蛋白沉积。因此,VILI和氧通过常见的急性肺损伤使肺损伤机制和可能协同或分析(图1)。lung-protective通风策略发展的关键是确定一个最佳的平衡补充啊2治疗和机械通风可以实现最小化的有害影响。

胞内炎症通路的示意图表示说明氧过多和机械肺损伤过度拉伸或剪切使用相同的细胞内途径启动炎症级联。NF-κB =核转录因子k B;PAI-1 =纤溶酶原激活物inhibitor-1;信使rna信使核糖核酸。查看详细描述文本。
VILI和氧急性肺损伤的影响
几个额外的动物模型发现,要么初加工的肺氧过多之前高弹潮汐卷(VT)或结合氧过多与高弹通风显著放大VILI的程度。24,26- - - - - -36这些模型模拟常见的临床策略管理ARDS和其他形式的急性呼吸衰竭出现之前lung-protective通风。总之,与控制相比,high-VT通风环境FIO2(0.21),或生理VT与氧过多high-V的结合T(18 - 30毫升/公斤)通风,和氧过多(FIO2= 0.8 - -1.0)显著增强VILI无数符号,包括:altered-permeability肺水肿的形成,24,26,27,31日,32弥漫性间质和肺泡出血,33,34降低表面活性剂生产(图2),28和肺的依从性,28,32,33炎性介质表达增加(图3)24,26- - - - - -28,31日,36以及细胞凋亡增加,26,33,34和增加肺泡由中性粒细胞浸润。24,26,27,31日- - - - - -33,36也发现了类似的结果,即使中等氧过多(FIO2结合V = 0.5)T25毫升/公斤。29日尽管上面列出的几个研究使用一个新生儿的影响31日,32或成年动物模型,24,26- - - - - -29日病理结果相似。


促炎症介质浓度从支气管肺泡灌洗(BAL)流体取自动物暴露于高弹潮汐通风的氧浓度21或90%。水平的肿瘤坏死因子α(TNF-α)明显高于过度拉伸条件下加氧过多,似乎一个互动效应(A)。另一方面,白细胞介素- 6 (il - 6)水平明显高于控制条件,但似乎没有出现增强彼此的炎症效应(B)。*P< . 05与无伸缩组21%的氧气浓度。从参考28许可。
此外,在主题与low-V ARDS管理T防护通风,长期(平均17 h,四分位范围8-33 h)不必要的接触相对较高IO2尽管有足够的氧化(FIO2> 0.50的年代阿宝2> 92%)与恶化氧化指数在48 h剂量反应的方式。38超过50%的相对氧与F队列进行管理IO20.70 >,> 70%的研究样本是管理41%的第一个48 h ARDS的过度FIO2(∼20 h)。氧组有明显机械通风和ICU停留时间的延长,尽管死亡率没有不同。尽管明显的数学之间的联系IO2和氧合指数,这些结果表明,长时间暴露于氧过多可能导致肺功能障碍的急性肺部炎症。
其他因素也会影响VILI和氧急性肺损伤。在某种程度上,损伤似乎是由成年或老年动物是否检查。尽管6毫升/公斤VT,老年大鼠暴露于FIO21 3 - 6 h遭受更大的恶化氧化和比成年鼠急性血碳酸过多症。35这些发现也与更大的肺毛细血管渗漏,促炎细胞因子,增加活性氧水平与细胞膜损伤和嗜中性粒细胞的激活有关。这可能反映了一个事实,寿命,累计氧化损伤必然导致变性的DNA,蛋白质,和其他大分子加剧急性ROS损伤或反映了衰老的抗氧化防御机制减弱生物基因损害。估计的氧化数支安打是暂时性的每个细胞的DNA损害约10000 /单元/ d对人类和老鼠100000 /单元/ d。39这反映了一个事实,氧化损伤是有氧代谢水平紧密联系在一起(这是在老鼠比人类高出7倍)。39这也解释了加速VILI和氧急性肺损伤的影响相比,小型哺乳动物与人类,为什么推广临床前研究的结果对人类必须保持谨慎,至少对于速度和氧化损伤的严重性。
最有趣的发现是,肺泡上皮细胞培养暴露于氧过多48 h(0.8 - -0.9)和过度紧张引起的病态的改造和重组的细胞骨架。30.氧过多加强了细胞膜,增加了其在模拟潮汐拉伸变形阻力。随着细胞被附加到一个人工基底膜的引入潮汐应变为20%(5倍估计正常VT应变)导致实质性的超然的肺泡细胞支持矩阵。研究者推测,防止损伤,变形特征的肺泡细胞和细胞外基质(他们是连接通过细胞骨架之间的联系和整合蛋白)40应该近似。肺泡上皮细胞的膜柔软(相对于基底膜)氧化应激似乎引起剪切,提高stretch-induced受伤。
临床前证据,使用几个不同的物种,清楚地表明,氧过多放大VILI的影响引起的机械部队在机械通气。这些模型使用一个相当高的VT在动物与正常肺。在实验中,使用了一个生理VT(7毫升/公斤)相比,氧过多的有害的影响通常是短暂的学习时间内未见(通常4 - 5小时)。24,27然而,VILI的历史研究在这方面很有启发性。韦伯和蒂尔尼的重要性13经典研究VILI最初并不受欢迎,因为他们的模型是基于正常肺通风非常大VT不用于临床实践。临床相关性才明显一旦意识到在ARDS肺损伤是不均匀分布的,所以常用的VT水平的12 - 15毫升/公斤功能相当于40毫升/公斤的成年正常充气肺组织接近5-y-old的孩子。41
因此,VT大小是一个相对的因素产生VILI基于多孔组织的数量和分布。区域与氧肺恶性通货膨胀气体可能强化组织损伤。这是以Terragni等,42报告小组的中度ARDS,其中很大一部分的V 6毫升/公斤T是优先分配给overdistended地区。尽管生产一直被认为是一个保护高原压力(29厘米H2O),这些受试者对炎性介质水平较高的支气管肺泡灌洗液与那些VT分发给正常充气组织(一个相应的高原压力H 26厘米2O)。事实上,那些通风是分布式overdistended地区也加重和通风不良FIO2(0.8和0.56)。42此外,需要注意的是,ARDS的放射检查正常的肺的地区也显示显著的炎症活动的证据。43因此,外观正常的肺在ARDS是具有欺骗性的。这些少或者最低限度受损组织仍然容易受到进一步破坏区域恶性通货膨胀和氧化损伤的影响。
长期补充氧气治疗慢性肺部疾病的影响
在1970年代早期,小等44报道,几乎一半的受试者与慢性阻塞性肺病长期O2治疗(∼2 y估计FIO20.22 - -0.27)的经典研究O2毒性解剖考试(即毛细血管增生,间质纤维化,上皮增生)。虽然这个发现似乎与临床前研究的影响,现在越来越多的证据表明,一个更复杂的病理生理过程的氧化损伤患者人群。慢性阻塞性肺病患者遭受氧化应激相关的慢性炎症长期暴露于香烟烟雾和传染性急性加重。45主题与慢性阻塞性肺病已经发现一种蛋白质硫醇损害抗氧化防御不足,所以,即使是短期的(18-48-h)补充O2在2 L / min放大ROS生产。46然而,正如其他人所注意到的那样,氧化应激在慢性阻塞性肺病病人停止吸烟后持续很久。45这表明,其他因素,如羰基二次压力(高活性有机分子的形成二次氧化应激的长期损害),在保持肺损伤中发挥作用。45此外,环境污染也会产生许多呼吸道进入肺部的氧化剂和其他组织,产生活性氧。COPD患者尤其容易受到这种环境的氧化损伤。46- - - - - -49因此,O的贡献2治疗不容易解析从其他因素。无论如何,补充O的好处2治疗严重的慢性肺部疾病患者远远大于氧化损伤的附加险。
系统性的影响氧过多
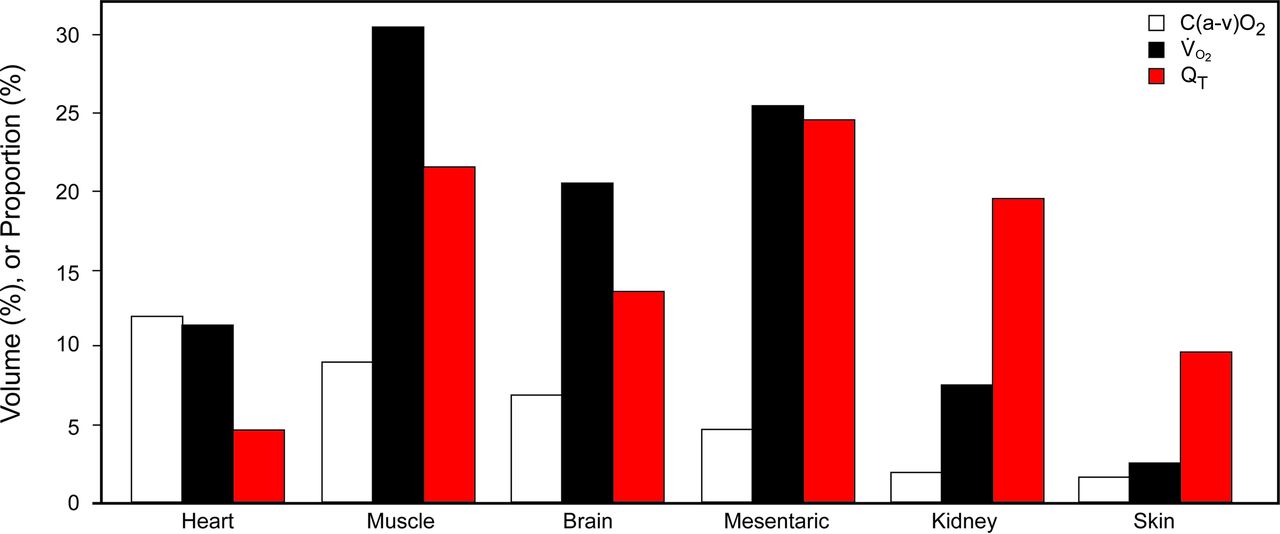
一直认识到,尽管肺成为第一个器官严重受到氧过多,大范围的损害发生远处器官显然取决于当地的灌注和代谢率。50基础代谢率,最高的器官2消费和灌注心脏、肌肉、大脑、腹部脏器(图4)。51已经观察到氧过多”会产生进步的细胞损伤和死亡在一个又一个器官系统,直到这个过程是停在肺损伤或死亡的动物”。50

描述oxygen-consuming最高的灌注和休息条件下体内器官。氧过多的有害影响内脏的明显受到代谢率和灌注。尽管肾脏,乍一看,似乎是在相对较低风险基于耗氧氧中毒,这是骗人的,考虑到高份额的心输出量。C(动)O2= arterial-venous氧含量不同;V̇O2=身体耗氧量的总量的百分比;问T=全身血流量的百分比。引用的数据50。
关于氧过多的系统性影响最突出的问题是,O2诱发系统性血管收缩,减少心输出量,降低对大多数组织灌注床,包括大脑、心脏、骨骼肌和皮肤。52减少灌注是线性和P成反比aO2。全身灌注似乎减少当PaO2超过150毫米汞柱,最大跌幅达到了20%。52这也许就是为什么提出截止临床上重要的动脉氧过多被一些人认为是一个PaO2> 150毫米汞柱。53
最令人信服的解释hyperoxia-induced血管收缩是ROS的产生超氧化物阴离子能灭活一氧化氮(NO):(1)减少l精氨酸(没有)的前体;(2)通过直接抑制酶没有合酶,或(3)由其效果作为配体,防止卸载的血红蛋白。52增加了不确定性,氧过多的血管收缩的影响可能是时间的性质,因为氧过多反而会增加l精氨酸合成酶。因此,氧过多的有害的影响,尤其是在缺血后再灌注损伤,可能需要考虑的性质和时间缺血或创伤。
允许氧过多患者的各种医疗条件已成为双方关注的领域,虽然高层普遍缺乏证据。52- - - - - -58总之,有明确的证据表明在正常受试者血管收缩反应氧过多存在剂量依赖的相关性,可以观察到在几分钟内,导致局部灌注平均减少30%。54回顾性研究的受试者post-cardiac被捕后的回归自然循环,持续暴露在动脉氧过多与贫穷有关神经结果和医院死亡的风险增加。53,56,57氧过多也被用于治疗急性脑损伤,其中大脑缺氧导致继发性脑损伤。55虽然有些病人似乎受益于氧过多,结果喜忧参半,仍然是有争议的话题。特别关注的是ROS的毒害神经的效果。氧化损伤脑组织和高死亡率增加3 - 6 h后氧过多暴露与脑缺血在动物身上得到证实。55在严重创伤性脑损伤,氧过多矛盾的影响区域的脑灌注高危组织(< 20毫升/ 100 g /分钟),也导致了至少改善脑组织PO2相比之下,受伤的地方。59
缺血再灌注损伤是O2悖论,即重建与含氧的血液灌注后缺血性事件矛盾导致细胞挛缩和坏死。60机制导致初始和后续的受伤是ROS的产生。因此,主要问题在很多情况下涉及危重病人氧过多的潜力来放大损害导致的缺血再灌注损伤。
总之,缺血触发一个氧化破裂通过诱导烟酰胺腺嘌呤二核苷酸磷酸和黄嘌呤氧化酶释放。这反过来又降低了啊2成超氧化物阴离子,破坏细胞膜,造成进一步的活性氧产量。因此,缺血组织成为准备维持进一步损害的逆转缺血。1,60,61年组织启动至少部分损耗引起的细胞内的抗氧化防御系统在最初缺血性事件。60在再灌注和氧合血,ROS生产进一步刺激,通过O的存在2和炎症级联,在正常组织损伤。活性氧产量成正比局部组织PO2,1所以氧过多进一步增加活性氧的生产和放大炎症减少抗氧化防御系统的上下文中。这全球的后果,因为炎症级联由缺血再灌注损伤导致远程损伤其他器官系统。61年
越来越多的临床和临床证据表明,氧过多在心肺旁路,58心脏骤停后,62年肝缺血,63年脑缺血,64年引起多器官损伤,表明氧过多应该尽可能避免。心脏骤停的临床前研究的荟萃分析65年发现复苏FIO2(1)持续60分钟回归后的自然循环生产大大增强神经元损伤及神经功能缺损严重而FIO20.21或维持正常动脉氧合滴定。泛化的结果从临床前试验,临床实践是很有问题;因此,对改变啊2政府在复苏期间不能推荐。然而,避免氧过多post-arrest期间为了减少伤害与缺血再灌注损伤是可行的。氧过多发现在心肺复苏后的ICU有更高的死亡风险而normoxia(优势比为1.8 (95% CI 1.5 - -2.2),P< . 01)。66年事实上,与氧过多与受试者的死亡率明显高于那些血氧不足(6%的比例不同,P< . 01)。
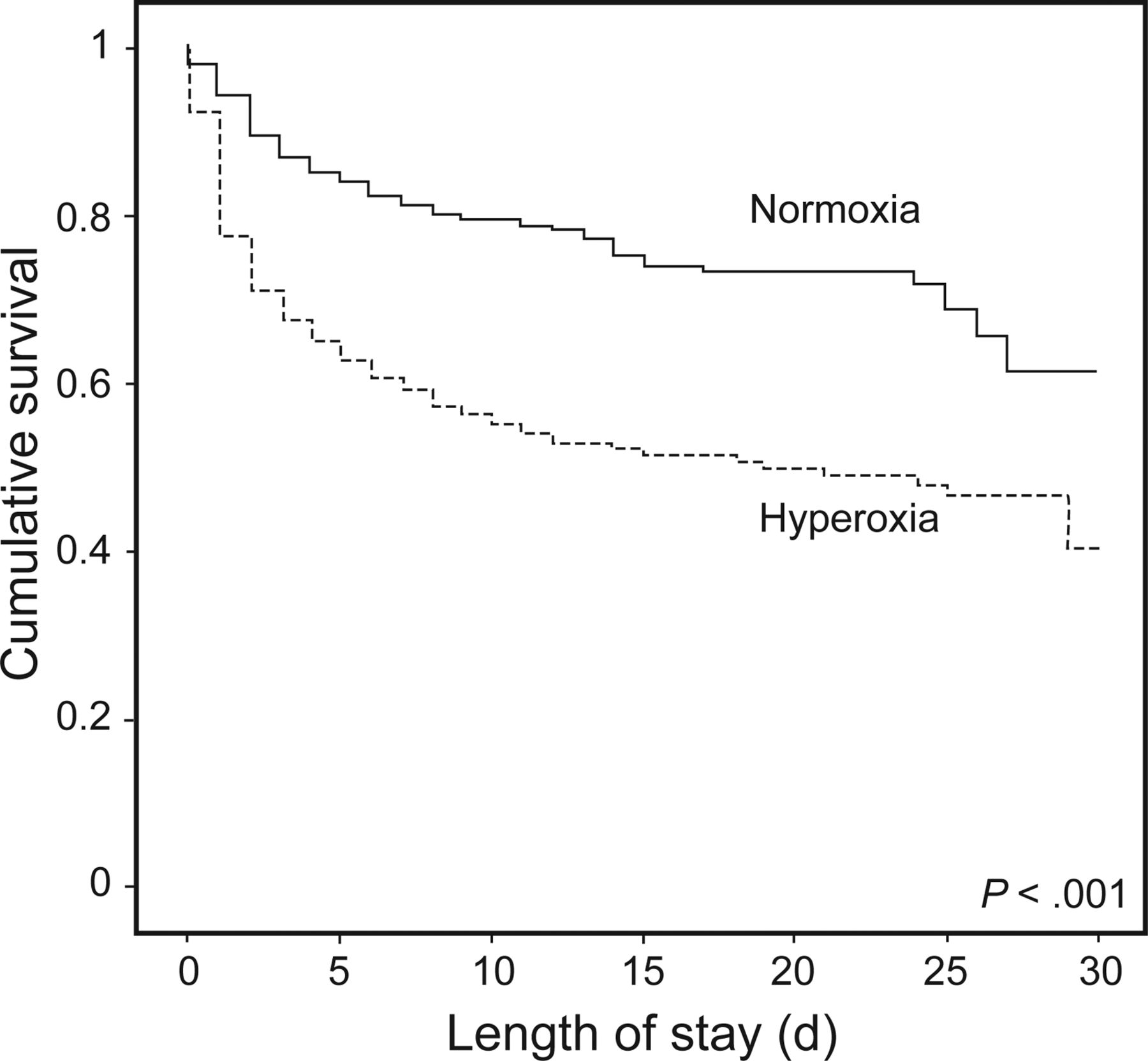
类似的医院死亡率增加的风险暴露于氧过多(PaO2≥300毫米汞柱)急性脑损伤后缺血性中风也有报道。67年氧组的住院死亡率为60%与53%相比那些暴露于低氧血症(PaO2< 60毫米汞柱),47%在这些分类是常氧。为其他混杂变量调整后,死亡的几率比氧过多组要明显高于normoxia(优势比为1.7 (95% CI 1.3 - -2.1),P<措施)以及那些暴露于低氧血症(优势比为1.3 (95% CI 1.1 - -1.7),P< . 01)。同样,使用相同的分类模式,创伤性脑损伤后暴露于氧过多也被独立医院更高的死亡率有关而normoxia(调整后的优势比为1.5 (95% CI 1.02 - -2.4),P< .04点)(图5)。68年

kaplan meier存活曲线说明了生存在创伤性脑损伤患者暴露于动脉氧过多。类似kaplan meier曲线已经证明当缺血性中风病人和post-cardiac逮捕也暴露于动脉氧过多。从参考63年许可。
此外,允许氧过多患者的院前和急诊科护理中慢性阻塞性肺病患者急性恶化与更高的呼吸性酸中毒发生率和机械通风的必要性以及医院死亡率增加。69年,70年在这些研究中,干预组O2治疗滴定实现S阿宝288 - 92%。与死亡率相关似乎没有氧过多本身而是高碳酸血症和呼吸性酸中毒的继发效应。血碳酸过多症是独立与慢性阻塞性肺病患者的死亡率有关,71年然而,目前尚不清楚该协会在流行病学研究发现仅仅是一个更严重的疾病的象征。无论如何,证据证明之间的关联hyperoxia-induced呼吸性酸中毒的患者的院前死亡率与医院急诊科设置提出了一个更为严重的过程。例如,在慢性阻塞性肺病急性呼吸性酸中毒患者肺动脉高压可能恶化或诱发肺心病。
最后,36000年一项研究涉及>机械通风学科从50 icu在荷兰,PaO2和FIO2在第一个24小时的机械通风与死亡率增加有关。72年当分析作为一个连续变量,最初增加PaO2与医院的死亡率下降,预计将与逆转的严重低氧血症,然后第二个死亡率增加似乎发生一次PaO2开始增加超过150毫米汞柱。在多元回归模型调整其他并发症,死亡率之间的关系,PaO2和FIO2依然存在。这些结果显示(但不证明),肺循环和体循环氧过多可能产生负面影响死亡率通过本文前面描述的机制。此外,研究结果强调临床管理策略的重要性,防止低氧血症同时最小化氧过多的发病率。
宽容的血氧不足作为一个策略来控制氧过多
越来越多的证据表明暴露病人氧过多是有害的,不明显的方式在日常临床实践。这导致了管理应该允许的提议宽松的血氧不足。这个策略以坚持严格的参数血红蛋白浓度(9 - 10 g / dL)和药物诱导异常的心脏指数(> 4.5 L / min / m2)维持正常组织O2交付。与这些警告,宽容的血氧不足允许患者管理一个PaO250 - 60毫米汞柱。73年尽管存在孤立的报道ARDS患者伴有严重低氧血症(即PaO2< 30毫米汞柱)没有证据表明组织缺氧,74年的误差是非常狭窄的患者容易受到意外的急性稀释或血液动力学不稳定。此外,在药物心脏工作量增加的背景下,重要的是要强调数据建立的有害影响血氧不足在ARDS和其对心脏功能与死亡率增加。75年,76年
一个发人深省的教训来自宽容血氧不足的早期测试管理二早产儿在促进和支持试验。77年,78年这些试验的O2治疗,干预组滴定来维持阿宝2在85至91年间,89%与95%之间。在支持试验中,早产儿(周妊娠)24日随机到较低的年代阿宝2管理部门有一个更高的死亡率分别为19.9%和16.2%,P= .04点)。77年死亡率同样高的低阿宝2群提高II试验(23.1% vs 15.9%,P= .002)。78年其他人指出,79年不存在明确地接受,降低组织氧阈值,可以容忍的。未来技术和生物标志物可以宽容的血氧不足。但是现在听起来似乎没有理由将这种策略引入到临床实践。
不论合法问题用这种方法,宽松的血氧不足的概念是有益的,它提醒人们,大多数患者可以管理一个PaO260 - 80毫米汞柱之间。这就提出了一个问题关于两个潜在的坏习惯在临床实践中。第一个是让患者管理持续的年代阿宝2没有验证的98 - 100%相应的PaO2。血红蛋白的亲和力2降低PaO2超过95毫米汞柱,PaO2当P饱和度通常达到100%aO2大约是250毫米汞柱。80年减少对阿2作为血红蛋白的方法完全饱和,加上固有的局限性脉搏血氧仪在检测动脉氧过多,意味着当年代阿宝2> 95%,小增加氧化检测到S阿宝2可能发生非常大的变化在PaO2。81年
第二个潜在的坏习惯是保持超常PaO2(特别是在有毒的F水平IO2)提供一个缓冲或安全边际急性稀释。O2等离子体的承载能力是微不足道的(P 0.003 mL / dL /毫米汞柱aO2)与血红蛋白(1.39 mL / g / dL)。82年临床医生可能会被误一种虚假的安全感保持超常PaO2患者的氧合状态。然而,它提供了一个相当微不足道的O2交付的缓冲区,一个微不足道的影响的增加静脉血掺杂。因此,维持某种程度的动脉氧过多的整体风险对冲PaO2稀释可能比小的潜在好处。例如,增加了PaO2从100到150毫米汞柱(重要动脉氧过多的尖端)增加O2交付能力的循环血容量(如5 L)估计有15毫升,或< 2%(假设一个正常的氧合血红蛋白曲线与相应增加aO2从98%到97)。认为需要有毒水平的FIO2(≥0.70)允许PaO2缓冲区(PaO2100毫米汞柱)的意义是基于现有的证据,因为潜在的危害大于任何感知心灵的平静可能给临床医生。
同样,维护动脉氧过多的病人遭受冲击peri-resuscitation期间矛盾可能导致弊大于利,放大缺血性再灌注损伤。话虽这么说,显然瞬态氧过多在复苏表示(因为缺氧的风险大于潜在风险从氧过多给我们目前的知识水平),是使用瞬态过程如插管期间氧过多,在维护一个肺啊2储备是明智的,以确保患者安全的事故或窒息时期的困难。
摘要职业的论点
总之,人类进化的演化线有极其敏锐的能力适应缺氧,但像其他哺乳动物一样,并不是特别具有强大的抗氧化防御机制在细胞水平来应对严重的氧化应激。氧急性肺损伤造成长时间暴露于有毒的氧(FIO2≥0.70)是一个非常有效的关注管理急性呼吸衰竭患者,因为它可能加剧了潜在的炎症过程,需要机械通气以及VILI。但也有证据表明内部器官(免受大气O2)可能更容易受到低水平的浓度比肺氧过多,该漏洞放大了其代谢率和灌注。2016年,谨慎的做法是,宁可谨慎采用通风机和辅助治疗策略,防止氧急性肺损伤和VILI。这些策略包括避免P的一部分aO2> 100毫米汞柱。采用美国国立卫生研究院的ARDS净37参数PaO2(55 - 80毫米汞柱)和S阿宝2(88 - 95%)出现在这方面特别有吸引力,因为这提供了一个合理的范围内适当的氧化和可以帮助临床医生为了避免或减少相关的风险与肺循环和体循环氧过多。
F的反对严格控制IO2
毫无疑问,氧气是一种有效的药物,是一种最常见的药物在紧急和急救护理。这通常是复杂的事实,氧气是无需处方,在一个未知的剂量,没有预定义的结束点。83年- - - - - -85年氧气可以挽救生命,但是氧气也是有毒的。氧气是一种生物活性分子,在宿主防御和调节细胞内信号通路以及氧化应激。86年氧气是促炎和抗炎。这个生命的双原子分子的二元性是讽刺,但讲述了氧气的复杂本质的人类生理学(表1)。作为职业中描述参数,氧气毒性的动物模型是众所周知的,并进一步讨论的观点是毫无根据的。16- - - - - -35这里的论点是:我们应该使用保守的氧疗法和避免hyperoxemia在机械通气病人吗?
氧气过剩的风险不容忽视,但也不应被夸大。重要的是要注意,氧过多从未发生。允许的电化学氧气的发现成为可能的医疗进步和释放氧气的毒性。正如Severinghaus和Astrup提到的,87年如果今天介绍了氧气作为药物,FDA不太可能会批准其使用。
氧气和通风的患者的结果
在过去的十年里,8个临床试验试图评估目标和hyperoxemia氧对结果的影响在机械通风38,71年,88年- - - - - -93年随着2荟萃分析。94年,95年其中4评估使用所谓的“保守氧气疗法。”89年,91年- - - - - -93年这里将详细考虑为了证明什么意见证据支持。每个试验中所示的一些显著的特点表2。
氧气疗法的实践在机械通气,当临床医生,是基于长期以来相信FIO2< 0.60是无毒的。事实上,早期应用程序经常偷看的引导与试图减少FIO2无毒的水平。氧气疗法一般也是提供,这样阿宝2足以提供一个缓冲的呼吸恶化,防止血氧不足。通过保持PaO2上平面的一部分氧合血红蛋白分解曲线,患者可以忍受突然改变肺功能没有血氧不足。在许多情况下,这将导致患者阿宝2> 96%。然而,这种做法也可以被视为掩蔽没有监控预警伤害严重恶化。此外,液态氧源在医院就足够了,这样节省氧气并不是一个问题。
Hyperoxemia和临床医生反应是在荷兰的审判96年的影响,评估FIO2设置和由此产生的年代阿宝2。这些研究人员发现,当FIO2> 0.60和氧过多(PaO2> 120毫米汞柱),临床医生反应是减少FIO2在大约80%的情况下。然而,当hyperoxemia时看到FIO2≤0.40,FIO2在只有四分之一的情况下降低。本研究> 5000例,> 120000血液气体样本似乎代表F的常见方法IO2世界各地。本文还提供了动力hyperoxemia的影响的研究。
在早期的生理研究高FIO2,Aboab等90年F相比IO20.6和1.0的5到14厘米H的窥视2为了确定阿高F的影响IO2在吸收肺不张。在连续14个科目PaO2/ FIO2< 300年,他们发现,呼吸气体FIO21.0与de-recruitment由于吸收肺不张。虽然这是一个优雅的研究,但结果并不奇怪,事实上并不告诉我们有用的信息关于这个问题。97年很明显,肺氧条件创建著名的肺功能障碍,但这并没有解决hyperoxemia。
在后续研究中,同样的荷兰研究人员评估回顾hyperoxemia对死亡率的影响,观察研究5 icu。71年3000年一群>的主题,他们找到了一个u型P之间的关系aO2在第一个24小时和死亡率。具体来说,最低和最高的科目PaO2有最大的死亡率,而F之间有一个线性关系IO2和死亡。他们还证实了以前的发现,受试者在这些icu往往PaO2高于文献中推荐值。很明显,这个试验表明,高FIO2低和高PaO2在第一个24小时与住院死亡率有关。这个试验证明因果关系。结果很容易可以解释疾病的严重程度。患者血氧不足耐火材料与氧气治疗可能,事实上,有这样严重的病理死亡是预期。同样,高FIO2是肺损伤的严重程度的标志,心脏功能,和需要的支持,又只是一个代理功能障碍的程度。
一项研究从澳大利亚和新西兰社会重症监护临床试验网络(ANZICS)回顾性评估动脉,最糟糕的肺泡梯度在第一次入住ICU的24小时150 ICU 9 y。通过多变量分析,他们试图确定P的影响aO2在死亡率。88年为网站和生理变量调整后,他们发现了一个血氧不足和结果之间的关系而不是hyperoxemia和结果。他们得出结论,在机械通风ICU主题,hyperoxemia的角色在最好的结果是不确定的。
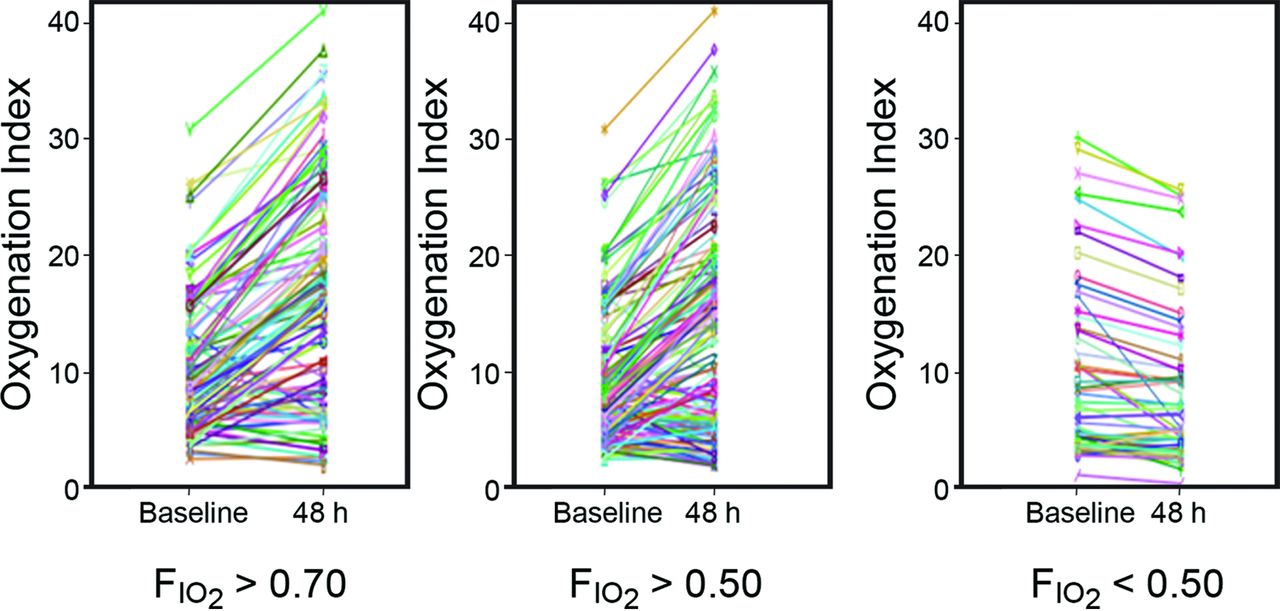
梅奥诊所的研究是第一个研究暗示可能氧过多的负担和氧气接触住院肺功能。38这组评估FIO2和相应的页aO2在210例48 h的通气支持。他们过度F定义IO2> 0.5,而S阿宝2是> 92% (图6)。过度的负担IO2与氧合指数恶化在48 h剂量依赖性的方式(图7)。在这些学科的最大FIO2负担,一个协会天在机械通气,ICU停留更长,和更长的住院时间。然而,有没有对死亡率的影响。与ANZICS审判88年,这些发现可能归因于疾病的严重程度。然而,肺过度的接触氧气缺乏因果关系需要的是一个合理的理由。但这里的论点是死亡率,举证责任没有被满足。

过度的计算FIO2在一个烧焦的病人负担。从参考37。

改变氧化指数从起始的机械通气(基线)插管后48小时,与FIO2。从参考37。
保守的氧疗法试验
铃木等89年,91年评估使用保守的氧气治疗前后2试验。保守的氧疗法的定义是为了进一步讨论。根据伊斯特伍德等,98年保守的氧气治疗目标FIO2最低的90 - 92%的FIO2。尽管偷看氧化管理不可分割的一部分机械通风病人,偷看没有参与这个定义。
在第一次试验中,105名被试进行了研究,51在标准治疗,54后切换到保守的氧疗法。89年在标准治疗,意味着年代阿宝2是98%,而在保守的氧气疗法,年代意味着什么阿宝2是95%。作者得出的结论是,保守的氧气治疗是可行的和自由的不良临床或生化结果。这项研究并没有试图确定死亡率的差异。的二次分析同一组受试者,他们决定平均肺不张的分数和时间之间的第一个自主呼吸试验组。91年保守的肺不张得分较低氧疗法组,和第一次自主呼吸试验时间缩短。的解释,你可以说保守氧疗法可能促进断奶通过防止进行性肺崩溃。但最终,这小群不允许死亡的讨论。
Panwar等92年评估使用保守的氧气治疗103例4澳大利亚icu。年代阿宝288 - 92%和> 96%的目标比较通气支持的持续时间。保守的氧疗法组实现所需的年代阿宝2在不增加的时间百分比的年代阿宝2< 88%。在这个实验中死亡率没有改变。作者得出的结论是,保守的氧疗法是安全的和可行的。
最近的试验由荷兰研究人员使用保守的逐步实现氧疗法在仍时间框架。93年在第一步中,S阿宝2目标是92 - 95%,而第二步利用决策辅助系统指导协议的依从性。试验的主要终点是氧化的目标实现。尽管较低的年代阿宝2目标,没有差别在缺氧发作的持续时间。Ventilator-free日子更保守的氧疗法2期平均增加了0.5 d。调整ICU和医院死亡率保持不变。
过度FIO2在其他条件
尽管这场争论仅限于患者需要机械通气肺损伤,有很多其他疾病恶化的氧过多。常见的在这些疾病是侮辱,缺血/再灌注疾病的发病机制中起着重要的作用。缺血后hyperoxemia创建一个完美的环境更容易通过活性氧伤害生产。这些被认为是短暂的。
心肌梗死/心脏骤停
尽管氧气被广泛规定心脏骤停后心肌梗死,氧过多的负面的生理效应已经知道持续很长一段时间。99年- - - - - -101年Kilgannon等63年已经证明氧过多发挥了更大作用心脏骤停后死亡率比缺氧。其他一些调查和荟萃分析102年- - - - - -107年在这个问题上一直发表后Kilgannon的工作。这场辩论一直对氧过多心脏骤停后,证据落在保守的氧疗法和预防氧条件。
创伤性脑损伤
创伤性脑损伤代表另一个情况下,氧过多可能是缺氧一样危险。大量的动物实验和临床研究表明氧化的负面结果的两个极端。脑灌注和氧具有重要影响血管舒缩的语气除了脑代谢的重要会议的要求。尽管数据并不引人注目的心脏骤停,hyperoxemia,面对正常的颅内压和脑组织氧,似乎负面后果。108年- - - - - -111年
中风
缺血性脑损伤后中风似乎也是受hyperoxemia的负面影响。这些数据通常包括那些不是机械通风,但恶化的风险在斯托克城缺血/再灌注损伤是引人注目的。64年,112年,113年林康和同事的荟萃分析64年结果表明氧过多的直接负面影响在机械通风学科后中风。事实上,作者得出结论:“在加护病房通风中风患者承认,动脉氧过多是独立与住院死亡而normoxia或缺氧。这些数据凸显了需要控制的研究通风中风病危人群的再氧化。在缺乏临床试验的结果,应该避免不必要的氧气交付在通风中风病人。”
结论
氧气早就知道会毒害人的肺,和一个FIO21.0与吸附肺不张,肺泡破裂,血氧不足。氧损伤其他器官系统已成为一个新领域的调查在hyperoxemia会导致负面的结果。保守的氧疗法针对normoxemia可以很容易地辩护没有大量的证据关于对死亡率的影响。外的一氧化碳中毒、减压病和气体栓塞,FIO21.0没有好处。未来的保守的氧疗法试验可能通过F的闭环控制IO2的问题,克服人类控制变量的年代阿宝2。
讨论
马里尼:
我发现两个演讲刺激。在富裕的Kallet说话,新陈代谢和F的乘数IO2为受伤的共鸣在实验室里我们发现多年来与其他VILI辅助因子。我说这么多我想在这里所有的旧帽子,但是你可以采取相同的有害的通货膨胀和模式匹配不同的血管模式,或者只temperatures-temperatures,不是VILI的新陈代谢和得到完全不同的表达式。我认为这是一个非常真实且重要的一点你了。第二件事是你没有提到格罗克特和马丁的工作1珠穆朗玛峰高海拔适应环境,项目,等等。我发现东西有趣,因为作为一个门诊医生我生命的大部分时间里,我将病人走进诊所和PO2非常低的水平。一个人我记得从西雅图有血气,我画了我自己在办公室里的20毫米汞柱。我在实验室搞砸了,喊了第二个,19日回来!我画了19、20和21双分析一切。,他只是感觉有点不舒服。关键是他被改编。我们知之甚少的适应及其潜力。如果我们有可靠的标记不宽容,我们也许能够减少FIO2不少。
布兰森:
这种宽容的血碳酸过多症或低氧血症基本上试图适应病人在ICU。我们必须有我们看到的中心区域,我们喜欢饱和度> 85%或无论你的偏好。如果在几天我们只接受89%接受87%,至85%,等等?显然我们担心当病人严重的血氧过低的直到他们在20厘米H2O窥视和FIO2是100%,PaO2是35或40毫米汞柱,一切都好。可能有一些适应环境,可以发生在危重病人。很明显,这需要更多的研究如何实施这一策略和其他因素而言,足够的心输出量,足够的血红蛋白,使发生在床边。
麦金太尔:
我做了一个期刊会议论文2在这几年前,引人入胜,约翰。最引人注目的变化之一,我不理解,你是一个胎儿,繁荣的PO2在30年代,然后交付的时候,它快速转换和P的生物会死O230。显然,有自适应机制,能够保持这种生物活着漂亮在子宫内,子宫内交货时大大改变。我们可以扭转,在ICU环境吗?我显然不知道答案。夏尔巴人和其他许多数周或数月显然适应并没有O去爬珠穆朗玛峰2。这使得他们PO2在30年代,甚至20年代。所以,显然有hypoxia-adaptive在我们的细胞机制。如何挖掘和激活他们和明智地使用它们是一个巨大的问号,但可能会有很大的潜力。
贝瑞:
氧是线粒体的最后一站。目标是保持几毫米汞柱的线粒体氧化线粒体PaO2。在高海拔或极端运动血氧不足,心输出量供应低氧含量增加氧气交付。因此,这对我来说是非常困难的理解研究不存在这些病人的心输出量和血液动力学。不仅是血液中的氧水平,但相关研究提供氧气和血液动力学
Kallet:
我认为回答这个问题的一件事不是夏尔巴人居住在喜马拉雅山和适应,但那些雅皮士爬珠穆朗玛峰。人不适应低氧血症。多长时间一个正常的人的攀登珠穆朗玛峰适应缺氧环境?
Mireles-Cabodevila:
数周甚至数月。
Kallet:
并不适用于危重病医学。
Mireles-Cabodevila:
困扰我的部分,关于血氧不足/氧过多在ARDS他们也有炎症过程。当你想到最关心的关于大脑的血氧不足的程度/氧过多。这些患者的长期结果肺损伤后,在性能方面,可能会影响到一个非常紧密的行为。这还有待研究。
Kallet:
有一些论文在ARDS的创伤后应激障碍的主题。一项研究3发现神经认知障碍1 y与长时间的血氧饱和度下降有关。有另一项研究中,我认为,在ARDS净,发现类似的结果在神经认知障碍之间1 y和氧化。4这真是一个烦人的问题。
Holets:
博士论文出来,Rachmale Pannu从梅奥诊所以抽象的形式在SCCM危重病医学(社会)therapist-driven协议使用电子警报系统来避免hyperoxemia病人。虽然这项研究是小和死亡率没有变化,附近有一个显著的趋势降低通风机天,ICU在干预组。
布兰森:
其实我相信ICUand氧过多是一个真正的问题,我们应该尽我们所能避免它。无论是F的闭环控制IO2或RT-led协议。让人们在40% O2用PO2150没有任何意义。
Mireles-Cabodevila:
除了你的弗吉尼亚州的与机械通气相关事件[]。
脚注
- 函授:理查德·H Kallet MSc RRT FAARC麻醉学系在旧金山加州大学旧金山总医院NH: GA-2,沿岸泥沙垅大街1001号,旧金山,CA 94110。
Kallet先生和布兰森先生提出了一个版本,本文在第54 RespiratoryC是日报》发布会上,“呼吸道护理争议三世”,于6月5 - 6,2015年,在佛罗里达州圣彼得堡。
布兰森先生与Mallickrodt披露关系,美敦力公司,明治医药、拜耳、Ventec。Kallet先生没有冲突披露。
- 版权©2016代达罗斯的企业
引用
- 1。↵
- 2。↵
- 3所示。↵
- 4所示。↵
- 5。↵
- 6。↵
- 7所示。↵
- 8。
- 9。↵
- 10。↵
- 11。
- 12。↵
- 13。↵
- 14。
- 15。↵
- 16。↵
- 17所示。↵
- 18岁。↵
- 19所示。↵
- 20.↵
- 21。↵
- 22。↵
- 23。↵
- 24。↵
- 25。↵
- 26岁。↵
- 27。↵
- 28。↵
- 29。↵
- 30.↵
- 31日。↵
- 32。↵
- 33。↵
- 34。↵
- 35。↵
- 36。↵
- 37岁。↵
- 38。↵
- 39岁。↵
- 40。↵
- 41岁。↵
- 42。↵
- 43。↵
- 44岁。↵
- 45岁。↵
- 46岁。↵
- 47岁。
- 48。
- 49。↵
- 50。↵
- 51。↵
- 52岁。↵
- 53岁。↵
- 54。↵
- 55。↵
- 56。↵
- 57。↵
- 58岁。↵
- 59。↵
- 60。↵
- 61年。↵
- 62年。↵
- 63年。↵
- 64年。↵
- 65年。↵
- 66年。↵
- 67年。↵
- 68年。↵
- 69年。↵
- 70年。↵
- 71年。↵
- 72年。↵
- 73年。↵
- 74年。↵
- 75年。↵
- 76年。↵
- 77年。↵
- 78年。↵
- 79年。↵
- 80年。↵
- 81年。↵
- 82年。↵
- 83年。↵
- 84年。
- 85年。↵
- 86年。↵
- 87年。↵
- 88年。↵
- 89年。↵
- 90年。↵
- 91年。↵
- 92年。↵
- 93年。↵
- 94年。↵
- 95年。↵
- 96年。↵
- 97年。↵
- 98年。↵
- 99年。↵
- One hundred.
- 101年。↵
- 102年。↵
- 103年。
- 104年。
- 105年。
- 106年。
- 107年。↵
- 108年。↵
- 109年。
- 110年。
- 111年。↵
- 112年。↵
- 113年。↵









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}